Microfluidic Nucleic Acid Extraction: A Comprehensive 2024 Guide for Research & Diagnostic Applications
This article provides a detailed overview of automated nucleic acid extraction (NAE) on microfluidic chips, targeting researchers and developers in biomedicine.
Microfluidic Nucleic Acid Extraction: A Comprehensive 2024 Guide for Research & Diagnostic Applications
Abstract
This article provides a detailed overview of automated nucleic acid extraction (NAE) on microfluidic chips, targeting researchers and developers in biomedicine. It explores the core principles and advantages of miniaturization, reviews current methodologies (magnetic bead, silica membrane, and emerging techniques), and addresses common optimization challenges. The guide concludes with validation strategies and a comparative analysis against conventional methods, highlighting the technology's transformative potential for point-of-care diagnostics, high-throughput screening, and next-generation sequencing workflows.
What is Microfluidic Nucleic Acid Extraction? Core Principles and Miniaturization Advantages
This application note details the technological evolution from conventional, macro-scale nucleic acid extraction kits to fully integrated microfluidic platforms. Framed within a thesis on automated nucleic acid extraction on a chip, this document provides comparative data, detailed protocols, and essential resource lists for researchers and development professionals aiming to transition to or develop microfluidic solutions.
Comparative Analysis: Macro-scale Kits vs. Microfluidic Chips
The transition involves significant changes in scale, reagent consumption, and integration. The following table summarizes key quantitative differences.
Table 1: Quantitative Comparison of Extraction Platforms
| Parameter | Conventional Macro-scale Kit (Column-based) | Integrated Microfluidic Chip |
|---|---|---|
| Typical Sample Volume | 200 µL - 1 mL | 1 µL - 100 µL |
| Total Reagent Consumption | 1 mL - 3 mL | 10 µL - 200 µL |
| Processing Time (Manual) | 45 - 90 minutes | < 30 minutes (automated on-chip) |
| Elution Volume | 50 - 200 µL | 5 - 20 µL |
| Final DNA Concentration | 10 - 100 ng/µL | 50 - 500 ng/µL (due to low elution vol.) |
| Throughput (Manual) | 1-12 samples per run | 1-96+ samples per chip (parallel) |
| Footprint | Bench-top centrifuge, heater, vortex | Compact instrument (<0.5 sq. m) |
Experimental Protocols
Protocol 1: Operation of a Typical Silica-Membrane Macro-scale Kit
This protocol is the benchmark against which microfluidic performance is measured.
1. Materials & Reagents:
- Sample: 200 µL whole blood.
- Commercial Kit (e.g., QIAamp DNA Blood Mini Kit).
- Ethanol (96-100%).
- Microcentrifuge.
- Heating block or water bath.
- Vortex mixer.
- Microcentrifuge tubes (1.5 mL, 2 mL).
2. Procedure:
- Lysis: Mix 200 µL sample with 200 µL lysis buffer and 20 µL Proteinase K. Vortex. Incubate at 56°C for 10 min.
- Binding: Add 200 µL ethanol (96-100%) to the lysate. Vortex. Transfer mixture to a spin column. Centrifuge at 6,000 x g for 1 min. Discard flow-through.
- Washing: Add 500 µL Wash Buffer 1 to the column. Centrifuge at 6,000 x g for 1 min. Discard flow-through. Add 500 µL Wash Buffer 2. Centrifuge at full speed (20,000 x g) for 3 min. Discard flow-through.
- Elution: Place column in a clean 1.5 mL tube. Apply 50-200 µL Elution Buffer (or AE buffer) to the center of the membrane. Incubate at room temperature for 5 min. Centrifuge at full speed for 1 min to elute DNA.
- Storage: Store eluted DNA at -20°C.
Protocol 2: On-Chip Nucleic Acid Extraction Using a Solid-Phase Reversible Immobilization (SPRI) Bead Method
This protocol details a common method adapted for microfluidic automation.
1. Materials & Reagents:
- On-Chip Materials: PDMS-glass hybrid chip with integrated magnetic micro-actuators.
- Reagents: Paramagnetic silica beads, Guanidinium-based lysis/binding buffer, 80% Ethanol wash buffer, TE elution buffer.
- Instrumentation: Custom microfluidic controller with pneumatic valves and syringe pumps.
2. Procedure:
- Priming: Hydrodynamically prime all chip channels with their respective buffers.
- Sample/Bead Loading: Introduce 20 µL of pre-mixed sample and paramagnetic silica beads in lysis/binding buffer into the designated chamber.
- On-Chip Incubation: Allow lysis/binding for 5 minutes at room temperature on-chip. Activate integrated magnetic actuators to immobilize bead-DNA complexes against the chamber wall.
- Washing: With beads immobilized, flow 100 µL of 80% ethanol wash buffer through the chamber over 1 minute. Deactivate magnets briefly, then reactivate to re-capture beads. Repeat with a second wash.
- Drying & Elution: Flow air through the chamber for 30 seconds to dry the beads. Deactivate magnets and resuspend beads in 10 µL of TE buffer (65°C). Incubate for 2 minutes. Activate magnets to immobilize beads, then transfer the eluted DNA to a clean outlet reservoir.
- Collection: The purified nucleic acid (now in ~8 µL) is collected via pipette from the outlet port for downstream analysis.
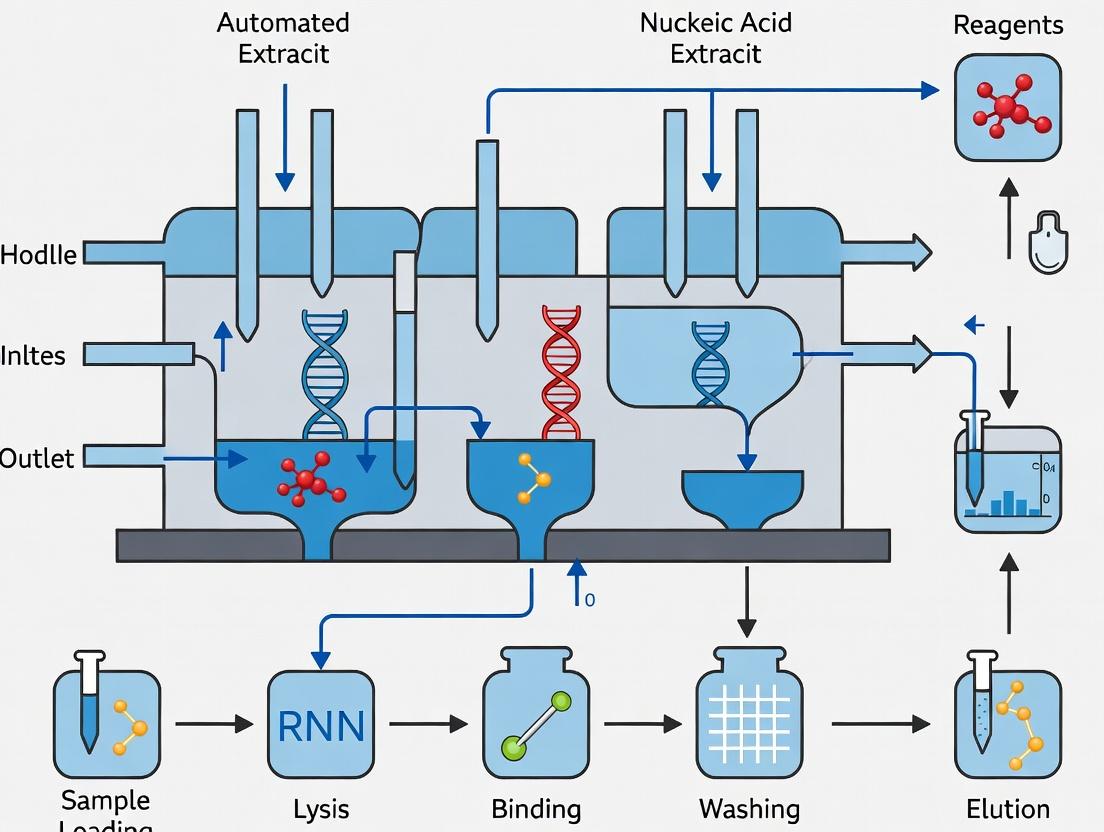
Workflow and System Diagrams
Title: Comparison of Macro vs. Microfluidic Nucleic Acid Extraction Workflows
Title: Functional Components of an Integrated Microfluidic NA Extraction Chip
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Microfluidic NA Extraction Development
| Item | Function in Microfluidic Context |
|---|---|
| Paramagnetic Silica Beads | Solid-phase matrix for nucleic acid binding; enables immobilization and movement via integrated magnets, eliminating need for centrifugation. |
| Guanidine Thiocyanate (GuSCN) Lysis Buffer | Chaotropic agent for cell lysis and DNA binding to silica surfaces; highly concentrated stocks used for on-chip dilution. |
| Surface-Active Agents (e.g., PEG, Triton X-100) | Added to binding buffers to enhance efficiency in low-volume, micro-scale mixing conditions. |
| PDMS (Polydimethylsiloxane) | Elastomeric polymer used to fabricate chip components via soft lithography; allows for integration of pneumatic valves. |
| Fluorinated Oil (for Droplet-based Chips) | Immiscible phase used to generate picoliter-nanoliter droplets for digital or single-cell extraction workflows. |
| SYBR Gold Nucleic Acid Stain | Fluorescent dye for real-time, on-chip quantification of DNA/RNA during or after extraction. |
| Bovine Serum Albumin (BSA) | Used to passivate microchannels and prevent non-specific adsorption of biomolecules to chip surfaces. |
This document provides application notes and experimental protocols for leveraging core physics principles of miniaturization—laminar flow, surface-to-volume ratio, and diffusion—within the framework of automated nucleic acid extraction on a microfluidic chip. Optimizing these principles is critical for enhancing extraction efficiency, purity, speed, and integration in downstream diagnostic and drug development applications.
Application Notes
Laminar Flow in Microfluidic Extraction
In microchannels (typically Dh < 500 µm), flow is characterized by low Reynolds numbers (Re << 2000), resulting in laminar, parallel streamlines without turbulence. This principle is exploited for precise reagent delivery, on-chip valving, and the creation of concentration gradients without mixing, except by diffusion.
Key Application: Laminar flow enables the "flow-over" technique, where a lysed sample stream flows adjacent to a stationary phase (e.g., silica-coated micropillars). Biomolecules diffuse perpendicularly to the flow to bind the surface, while cellular debris is carried away in the streamline, improving purity.
Surface-to-Volume Ratio (S/V)
Miniaturization drastically increases the S/V ratio. For a spherical droplet of radius r, S/V = 3/r. Scaling from a 1 mL tube (S/V ~ 60 cm⁻¹) to a 100 µm diameter channel (S/V ~ 4000 cm⁻¹) increases surface dominance.
Key Application: A high S/V ratio maximizes the effective area for solid-phase extraction (SPE). Silica-based binding surfaces can be engineered as micropillars, membranes, or beads packed in chambers, significantly increasing DNA binding capacity per unit volume and reducing reagent volumes for elution.
Diffusion as a Transport and Mixing Mechanism
At the microscale, molecular diffusion becomes a primary transport mechanism. The time (t) for a molecule to diffuse a distance x is approximated by t ≈ x²/2D, where D is the diffusion coefficient.
Key Application: Binding kinetics during nucleic acid capture are governed by diffusion to the surface. Efficient washing and elution require optimization of incubation times based on diffusive timescales. Rapid mixing for lysis or neutralization is achieved using chaotic advection geometries (e.g., serpentine channels) to reduce diffusive distances.
Table 1: Characteristic Parameters at the Microscale vs. Macroscale
| Parameter | Macroscale (1.5 mL Tube) | Microscale (100 µm wide channel) | Impact on Nucleic Acid Extraction |
|---|---|---|---|
| Reynolds Number (Re) | 100 - 1000 (Transitional) | 0.01 - 10 (Highly Laminar) | Predictable flow, no turbulent mixing. |
| Surface-to-Volume Ratio (cm⁻¹) | ~60 | ~4000 | Enhanced surface binding efficiency. |
| Diffusion Time for DNA (D ~ 10⁻¹² m²/s) | ~8.3 hours (to mix 1 cm) | ~0.5 seconds (to mix 100 µm) | Faster binding/washing if distances are small. |
| Typical Volumes Processed | 100 µL - 1 mL | 1 nL - 10 µL | Reduced sample/reagent consumption. |
| Heat Transfer Rate | Slow | Very Fast | Rapid thermal cycling for integrated lysis. |
Table 2: Diffusion Times for Key Molecules
| Molecule | Approx. Diff. Coeff. (D) in Water (m²/s) | Time to Diffuse 100 µm | Time to Diffuse 1 mm |
|---|---|---|---|
| Small Ion / Buffer Molecule | 1 x 10⁻⁹ | 0.005 s | 0.5 s |
| Protein (e.g., BSA) | 7 x 10⁻¹¹ | 0.07 s | 7.1 s |
| Genomic DNA Fragment (10 kbp) | 3 x 10⁻¹² | 1.7 s | 166.7 s (~2.8 min) |
Experimental Protocols
Protocol 1: Characterizing Laminar Flow Profile for Reagent Segmentation
Objective: To establish and visualize laminar co-flow streams for segmented delivery of lysis, wash, and elution buffers. Materials: See "Scientist's Toolkit" (Table 3). Method:
- Chip Priming: Place PDMS/glass microfluidic chip on microscope stage. Use syringe pumps to prime all channels with 70% ethanol at 5 µL/min for 10 minutes, then flush with DI water for 10 minutes.
- Flow Rate Calibration: Using two inlet pumps, introduce streams of food dye (Inlet A) and DI water (Inlet B) at equal flow rates (Q = 1 µL/min).
- Visualization & Measurement: Capture bright-field/video at 10x magnification at the confluence junction and 5 mm downstream. Use image analysis software (e.g., ImageJ) to measure stream width.
- Analysis: Confirm stable, parallel streamlines with a sharp interface. The interface width will broaden slightly downstream due to transverse diffusion.
Protocol 2: Optimizing Binding Incubation Time via Diffusion-Limited Capture
Objective: Determine the minimum residence time in a capture chamber for efficient DNA binding. Materials: See "Scientist's Toolkit" (Table 3). Method:
- Chip Preparation: Use a chip with a packed bed of silica-coated magnetic beads (50 µm diameter) in a 1 nL chamber.
- Sample Introduction: Flow a fluorescently-labeled DNA ladder (1 kbp-10 kbp, 0.1 µg/µL in binding buffer) through the chamber at a constant flow rate (Q = 0.5 µL/min).
- Time-Course Measurement: Use a confocal microscope to take fluorescence intensity snapshots of the chamber every 30 seconds for 10 minutes.
- Data Processing: Plot normalized chamber fluorescence (proxy for bound DNA) vs. time. Fit curve to an exponential rise model: F(t) = F_max(1 - e^{-kt}), where *k is the effective rate constant dominated by diffusion.
- Determination: The time to reach 95% of F_max is the optimal minimum incubation time for your chamber geometry.
Protocol 3: Evaluating Elution Efficiency via High S/V Ratio Effects
Objective: Assess elution volume and contact time needed for high-yield recovery from a high-S/V structure. Materials: See "Scientist's Toolkit" (Table 3). Method:
- DNA Capture: Load a known quantity of genomic DNA (e.g., 500 ng from lambda phage) in binding buffer onto the chip's SPE region (e.g., silica monolith). Wash with 10 chamber volumes of wash buffer.
- Elution Protocol: Apply a low-salt elution buffer (10 mM Tris-HCl, pH 8.5) in a stepwise manner. Collect five sequential eluate fractions (E1-E5), each equal to one chamber void volume.
- Quantification: Measure DNA concentration in each fraction using a fluorometric assay (e.g., Qubit).
- Analysis: Calculate cumulative yield. A well-designed high-S/V structure should release >90% of bound DNA in the first 2-3 void volumes, demonstrating efficient surface access.
Visualizations
Diagram 1: Microfluidic nucleic acid extraction workflow.
Diagram 2: Physics principles linked to chip effects.
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions & Materials
| Item | Function in Protocol | Example Product/Specification |
|---|---|---|
| PDMS Microfluidic Chip | Contains etched channels for fluidic operations. | Custom design with 100 µm x 100 µm channels, bonded to glass. |
| Programmable Syringe Pumps | Provide precise, pulseless flow for laminar regime. | neMESYS Low Pressure Syringe Pump. |
| Silica-Coated Magnetic Beads | High S/V solid phase for nucleic acid binding. | 1 µm diameter, superparamagnetic, sol-gel silica coating. |
| Chaotic Mixer Chip Design | Reduces diffusive path length for rapid mixing. | Herringbone or staggered herringbone micromixer. |
| Binding/Wash Buffer (High Salt) | Promotes nucleic acid adsorption to silica surface. | 6 M GuHCl, 10 mM Tris-HCl, pH 6.5, 0.1% Triton X-100. |
| Low-Salt Elution Buffer | Disrupts nucleic acid-silica interaction for release. | 10 mM Tris-HCl, pH 8.5, or nuclease-free water. |
| Fluorescent DNA Intercalating Dye | Visualizes flow streams and quantifies DNA. | SYBR Green I or YOYO-1. |
| Benchtop Fluorometer | Quantifies nucleic acid concentration in eluates. | Qubit 4 Fluorometer with dsDNA HS Assay. |
Application Notes: Microfluidic Nucleic Acid Extraction in Modern Research
Within the broader thesis on automated nucleic acid extraction on microfluidic chips, these core advantages converge to address critical bottlenecks in genomics, diagnostics, and drug development. The shift from conventional macroscale systems (e.g., column-based or magnetic bead processing in 96-well plates) to integrated microfluidic platforms fundamentally transforms workflow efficiency and accessibility.
Reduced Reagent Consumption: Microfluidic chips operate with nanoliter to microliter volumes, slashing reagent costs by 50-90% compared to standard protocols. This is paramount for expensive enzymatic mixes, specialized buffers, and novel therapeutic nucleic acid samples.
Faster Processing: By minimizing diffusion distances and enhancing surface-to-volume ratios, binding, washing, and elution steps are accelerated. Integrated automation allows parallel processing, turning multi-hour extraction and purification protocols into tasks completed in 10-30 minutes.
Enhanced Automation: On-chip valving, pumping, and fluidic routing, controlled by software, enable "sample-in, answer-out" operation. This minimizes manual intervention, reduces cross-contamination risk, and improves reproducibility for high-throughput applications like NGS library prep or pathogen screening in clinical trials.
Table 1: Performance Comparison: Microfluidic Chip vs. Conventional Benchtop Methods
| Parameter | Conventional Bench (Magnetic Beads) | Microfluidic Chip Platform (Representative) | Advantage |
|---|---|---|---|
| Sample Input Volume | 100-1000 µL | 10-100 µL | 90% reduction possible |
| Total Reagent Consumption | 500-2000 µL per prep | 50-150 µL per prep | 70-92% reduction |
| Total Processing Time | 60-120 minutes | 12-25 minutes | 4-5x faster |
| Hands-on Time | 30-45 minutes | <2 minutes (loading only) | Near-full automation |
| Elution Volume | 50-100 µL | 10-20 µL | Higher final concentration |
| Yield (from 200µL blood) | 60-85% | 70-90% | Comparable or improved |
| Parallelization | 8-96 samples (requires robotic handler) | 1-12 samples per chip (multiple chips runnable) | Simplified scalability |
Table 2: Impact on Downstream Applications
| Downstream Assay | Benefit from Microfluidic Extraction | Key Metric Improvement |
|---|---|---|
| qPCR / dPCR | Reduced inhibitor carryover, smaller elution volume. | Ct values reduced by 1-3 cycles; improved precision. |
| Next-Generation Sequencing (NGS) | Superior library prep efficiency from minimal sample. | Lower duplicate rates, better coverage uniformity from low-input samples. |
| Point-of-Care Diagnostics | Enables rapid, integrated systems for field use. | Time-to-result under 30 minutes from raw sample. |
| High-Throughput Drug Screening | Enables processing of thousands of cell culture/lysate samples. | Cost per sample reduced by >60% for genome-wide studies. |
Experimental Protocols
Protocol 1: On-Chip Solid-Phase Extraction Using Silica Membranes
Objective: Extract high-purity genomic DNA from whole blood on a pressure-driven PDMS/glass microfluidic chip.
Key Reagents & Materials: See "The Scientist's Toolkit" below.
Methodology:
- Chip Priming: Mount chip on controller. Flush all channels with 100 µL of nuclease-free water, followed by 50 µL of Binding Buffer (containing guanidine HCl and isopropanol).
- Sample Preparation & Loading: Mix 20 µL of fresh whole blood with 60 µL of Binding Buffer and 20 µL of Proteinase K off-chip. Incubate at 56°C for 5 min. Load the 100 µL lysate mixture into the designated sample reservoir.
- Automated Binding: Activate the on-chip pneumatic valves per the programmed sequence. The controller drives the lysate through the embedded silica membrane at a flow rate of 2 µL/sec. DNA binds to the silica under high-salt conditions.
- Washing: Sequential automated passage of 50 µL of Wash Buffer 1 (guanidine HCl, ethanol) and 80 µL of Wash Buffer 2 (ethanol, 70%) across the membrane at 3 µL/sec. Waste is directed to a separate reservoir.
- Elution: After a 30-second dry phase (air purge), elute purified DNA by passing 15 µL of pre-heated (70°C) Elution Buffer (10 mM Tris-HCl, pH 8.5) across the membrane at 1 µL/sec. Collect eluate from the output port.
- Analysis: Quantify DNA yield and purity via microvolume spectrophotometry and assess integrity by agarose gel electrophoresis or chip-based assay.
Protocol 2: Magnetic Bead-Based Extraction on a Digital Microfluidic (DMF) Chip
Objective: Perform automated RNA extraction from cell lysate using electrowetting-on-dielectric (EWOD).
Key Reagents & Materials: See "The Scientist's Toolkit" below.
Methodology:
- Chip Preparation: Load reagents (Lysis/Binding, Wash 1, Wash 2, Elution) into designated reservoirs on the DMF cartridge. Pipette 5 µL of functionalized magnetic beads and 25 µL of cell lysate into the reaction zone.
- Droplet Merging & Binding: Using software control, actuate electrodes to merge the bead and lysate droplets. Mix by moving the combined droplet back and forth for 3 minutes. RNA binds to beads under high-salt conditions.
- Bead Washing: Using an on-chip magnet, immobilize the bead pellet. Actuate electrodes to split the waste supernatant away. Move a 40 µL Wash 1 droplet over the beads, resuspend by droplet motion (1 min), and immobilize to remove waste. Repeat with Wash 2.
- Elution: Move a 12 µL Elution Buffer droplet over the washed beads. Mix for 2 minutes at 65°C (achieved by on-chip heater). Immobilize beads and move the purified RNA eluate droplet to the collection port.
- Collection & QC: Pipette the eluate from the port. Analyze using a bioanalyzer for RNA Integrity Number (RIN) and qRT-PCR for specific transcript recovery.
Visualizations
Diagram 1: Microfluidic NA Extraction Workflow
Diagram 2: System Architecture of an Automated Platform
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Microfluidic Nucleic Acid Extraction
| Item | Function & Specification | Example/Note |
|---|---|---|
| Microfluidic Chip | Disposable or reusable device containing microchannels, valves, and extraction media. | PDMS-glass hybrid with integrated silica membrane; or cartridge-based DMF chip. |
| Lysis/Binding Buffer | Chaotropic salt-based solution (e.g., guanidine HCl) to denature proteins and promote NA binding to solid phase. | Often contains RNA/DNA stabilizers and inhibitors of nucleases. |
| Wash Buffers | Ethanol-based solutions with decreasing salt concentrations to remove contaminants while retaining NA on solid phase. | First wash: chaotrope + ethanol. Second wash: ethanol-only for final cleanup. |
| Elution Buffer | Low-ionic-strength, slightly alkaline buffer (e.g., Tris-EDTA, Tris-HCl) to destabilize NA-solid phase interaction. | Heated to 65-70°C to increase elution efficiency. |
| Functionalized Magnetic Beads | Paramagnetic particles coated with silica or carboxyl groups for sequence-specific or total NA capture. | Size: 1-3 µm. Bead concentration optimized for binding capacity vs. handling. |
| Proteinase K | Serine protease included in lysis step to digest nucleases and other proteins, improving yield and purity. | Used for tough samples like tissue or blood. |
| Carrier RNA | RNA added to lysis buffer to improve recovery of low-concentration viral RNA or cfDNA by competing for tube/bead surface sites. | Critical for sensitive diagnostic applications. |
| Nuclease-Free Water | Used for preparing buffers and chip priming to prevent sample degradation. | Must be certified DNase/RNase-free. |
| Positive Control | Known quantity of NA (e.g., from cultured cells, synthetic oligos) spiked into negative matrix to validate extraction efficiency. | Essential for protocol optimization and QC. |
Within the context of automated nucleic acid extraction on a microfluidic chip, the fundamental differences between DNA and RNA as target molecules dictate distinct engineering and biochemical approaches. Successful on-chip integration requires a nuanced understanding of their contrasting physicochemical properties, stability profiles, and the consequent implications for protocol design. These considerations directly impact the efficiency, purity, and downstream applicability of the extracted nucleic acids for research and diagnostic applications.
Core Molecular Considerations: A Quantitative Comparison
The table below summarizes the key differentiating factors that must be addressed in microfluidic chip design.
Table 1: Critical Comparison of DNA and RNA for On-Chip Extraction
| Property | DNA | RNA | Implication for On-Chip Design |
|---|---|---|---|
| Chemical Structure | Deoxyribose sugar; Thymine base; Typically double-stranded. | Ribose sugar; Uracil base; Typically single-stranded. | RNA is more chemically labile due to ribose's 2'-OH group, requiring stringent RNase inhibition. |
| Stability | Highly stable; resistant to alkaline hydrolysis. | Labile; susceptible to hydrolysis and ubiquitous RNase degradation. | Chip surfaces/materials must be treated or selected to be RNase-free. Protocols must be faster or incorporate coolants for RNA. |
| Required Lysis Conditions | Robust; often uses alkaline lysis (e.g., NaOH) or strong ionic detergents (e.g., SDS). | More gentle; often uses chaotropic salts (e.g., GuHCl) combined with RNase inhibitors. | May necessitate separate lysis zones or reagent channels for DNA vs. RNA protocols on a universal chip. |
| Binding to Silica | Binds efficiently at high chaotropic salt concentrations (pH ≤ 7.5). | Binds efficiently at high chaotropic salt concentrations (with ethanol). Optimal binding at pH ≤ 7.5, but more sensitive to salt/ethanol ratios. | Similar but not identical binding chemistry. RNA may require more precise volumetric control of binding buffers. |
| Elution | Eluted in low-ionic-strength buffer (e.g., TE, Tris) or nuclease-free water at elevated temperature (65-70°C). | Eluted in nuclease-free water or TE buffer. Often eluted at 55-65°C to maintain integrity. | Separate thermal control zones may be needed. Elution reservoir must be certified RNase-free for RNA. |
| Common Downstream Use | PCR, sequencing, genotyping, cloning. | RT-qPCR, RNA-Seq, gene expression analysis, microarray. | RNA extraction purity is critical, as contaminants inhibit reverse transcriptase. |
Detailed On-Chip Protocols
Protocol A: On-Chip Genomic DNA Extraction from Cultured Cells
Application Note: This protocol is optimized for the isolation of high-molecular-weight genomic DNA from mammalian cells using a silica-membrane-based microfluidic chip.
Research Reagent Solutions & Materials:
| Item | Function |
|---|---|
| Chaotropic Lysis/Binding Buffer (e.g., GuHCl or NaI-based) | Denatures proteins, inhibits nucleases, and provides high-ionic-strength conditions for nucleic acid binding to silica. |
| RNase A (Optional) | Degrades contaminating RNA to increase DNA purity. |
| Wash Buffer 1 (Chaotropic salt + ethanol) | Removes contaminants while keeping DNA bound. |
| Wash Buffer 2 (Ethanol or ethanol-based buffer) | Removes salts and residual contaminants. |
| Low TE Buffer (10 mM Tris-HCl, 0.1 mM EDTA, pH 8.5) or Nuclease-free Water | Elutes pure DNA from the silica matrix. Stable pH of Tris preserves DNA. |
| Silica-Coated Microchamber | Solid-phase matrix for selective nucleic acid binding. |
| On-chip Pneumatic or Peristaltic Micropumps/Valves | Precisely controls fluid flow and timing. |
| Off-chip Centrifuge or Vacuum Manifold | Drives liquids through the silica membrane (for some chip designs). |
Experimental Workflow:
- Cell Loading: Introduce ~1×10^6 pelleted cells in PBS into the chip's input port.
- Lysis: Mix cell suspension with 200 µL of Lysis/Binding Buffer and 4 µL of RNase A (if desired). Incubate on-chip at room temperature for 2 minutes.
- Binding: Transfer the lysate across the silica-coated microchamber. Apply a negative pressure (or positive push) for 2 minutes to pass lysate through, enabling DNA binding.
- Washing: Pass 400 µL of Wash Buffer 1 through the chamber. Follow with 500 µL of Wash Buffer 2. Dry the membrane by pushing air through for 1 minute.
- Elution: Apply 50-100 µL of pre-heated (70°C) Low TE Buffer to the membrane. Incubate for 2 minutes. Collect the eluate by applying pressure. A second elution step can increase yield.
Protocol B: On-Chip Total RNA Extraction from Cultured Cells
Application Note: This protocol prioritizes speed and RNase inhibition for the isolation of intact total RNA, including miRNA, using a chaotropic-silica method.
Research Reagent Solutions & Materials:
| Item | Function |
|---|---|
| Denaturing Guanidine Isothiocyanate (GITC) Lysis Buffer with β-mercaptoethanol | Immediately inactivates RNases, denatures proteins, and disrupts cells. |
| Acid-Phenol:Chloroform | For phase separation; removes proteins and lipids (used in some chip designs with a separation zone). |
| Ethanol (70-80%) | Provides correct ionic conditions for RNA binding to silica and is used for washing. |
| DNase I (RNase-free) | Removes contaminating genomic DNA. |
| RNase-free Elution Buffer or Nuclease-free Water | Elutes RNA without degrading it. |
| RNase-free Silica-Coated Microchamber | Critical for preventing RNA degradation during binding. |
| Integrated Cooled Zone (4-15°C) | Maintains low temperature during binding/wash to further inhibit RNases. |
Experimental Workflow:
- Cell Loading & Lysis: Immediately lyse ~1×10^6 cells in 350 µL of GITC-based lysis buffer loaded onto the chip. Ensure complete homogenization within 30 seconds.
- Binding: Add 250 µL of ethanol (96-100%) to the lysate and mix. Transfer the mixture across the RNase-free silica chamber at 15°C. Apply pressure to pass the mixture through.
- DNase Treatment (On-Column): Prepare an on-membrane digest by applying 50 µL of DNase I incubation mix (in digestion buffer). Incubate on-chip at 20-25°C for 15 minutes.
- Washing: Wash with 400 µL of a low-salt buffer, followed by 500 µL of 80% ethanol. Dry the membrane with air for 1 minute.
- Elution: Elute with 30-50 µL of nuclease-free water (pre-heated to 65°C) by incubation and pressure-driven flow. Collect eluate in a nuclease-free tube.
Visualized Workflows and Considerations
Application Notes: The Integration of Automated NA Extraction in Microfluidics
The COVID-19 pandemic served as a profound catalyst, accelerating the development and deployment of point-of-care (POC) diagnostic platforms. This urgent demand underscored the critical need for fully automated, sample-to-answer systems, placing microfluidic-based automated nucleic acid (NA) extraction at the center of innovation. The evolution of Lab-on-a-Chip (LOC) technologies has transitioned from academic proof-of-concept to essential tools for rapid, decentralized testing. Within this thesis on automated NA extraction on microfluidic chips, the convergence of these three drivers is analyzed to define the specifications for next-generation systems: speed (<20 minutes), sensitivity (approaching PCR efficiency), and simplicity of operation for non-specialist settings.
Table 1: Performance Comparison of Recent Microfluidic NA Extraction Methods (2022-2024)
| Method (Chip Substrate) | Lysis Method | Capture Mechanism | Elution Volume | Efficiency (%) | Time (min) | Target Application |
|---|---|---|---|---|---|---|
| Silica-Membrane (PDMS) | Chemical (GITC) | Surface-functionalized pillar array | 15 µL | 85 ± 5 | 12 | SARS-CoV-2 RNA from saliva |
| Magnetic Bead (PMMA) | Thermal (65°C) | On-chip magnetic separator | 20 µL | 92 ± 3 | 8 | Bacterial DNA from whole blood |
| Electrokinetic (Glass) | Electrical (1.2 kV/cm) | Charge-selective trapping | 10 µL | 78 ± 7 | 5 | Viral RNA from nasopharyngeal swab |
| Aqueous Two-Phase (COP) | Chemical (Chaotropic salt) | Polymer phase separation | 25 µL | 70 ± 6 | 15 | Pathogen DNA from complex samples |
Experimental Protocols
Protocol 1: Integrated Magnetic Bead-Based NA Extraction on a Centrifugal Microfluidic Disk
This protocol details the automated extraction of viral RNA from a nasopharyngeal swab sample using a commercially available centrifugal microfluidic system (e.g., SpinDx-like platform).
Key Research Reagent Solutions & Materials:
| Item | Function |
|---|---|
| Lysis Buffer (Guanidine HCl + Triton X-100) | Disrupts viral envelope and inactivates nucleases. |
| Silica-Coated Superparamagnetic Beads | Bind nucleic acids under high chaotropic salt conditions. |
| Wash Buffer (Ethanol 70% v/v) | Removes salts, proteins, and other impurities while NA remains bead-bound. |
| Low-Salt Elution Buffer (TE, pH 8.5) | Provides optimal ionic conditions to release purified NA from beads into solution. |
| Wax Valves (Tridecane-based) | Thermally actuated to control fluid progression on the disk. |
| PCR Master Mix with Lyophilized Reagents | Pre-stored in detection chamber for downstream RT-qPCR. |
Procedure:
- Chip Priming: Load 200 µL of wash buffer and 50 µL of elution buffer into their respective reservoirs on the polystyrene disk. Pre-load the lysis chamber with 50 µL of lysis buffer and magnetic beads (10 µg).
- Sample Introduction: Pipette 200 µL of viral transport media containing the swab sample into the sample inlet port.
- Automated Run: a. Lysis & Binding: Spin disk at 800 rpm to propel sample into lysis chamber. Halt rotation for 5 minutes for chemical lysis. Resume rotation at 1500 rpm to mix lysate with beads and incubate for 3 minutes. Apply an external magnet to the chamber to immobilize bead-NA complexes. b. Washing: Spin at 2000 rpm while applying a moving magnet strategy to transfer beads sequentially through two stationary wash buffer chambers. c. Elution: Transfer beads to the elution chamber. Halt magnet, spin at 500 rpm to mix beads with elution buffer for 2 minutes. Apply magnet to immobilize beads, leaving purified RNA in solution. d. Distribution: Spin at 3000 rpm to meter 5 µL of eluate into the downstream RT-qPCR reaction chamber.
- Analysis: Transfer disk to a thermocycler reader for direct amplification and detection.
Protocol 2: On-Chip Electrowetting-on-Dielectric (EWOD) Digital NA Extraction for Single-Cell Analysis
This protocol enables the isolation and extraction of genomic DNA from individual cells for downstream sequencing, leveraging digital microfluidics.
Procedure:
- Device Preparation: Activate the EWOD chip (glass with patterned ITO electrodes) by applying a continuous hydrophobic coating (e.g., Cytop). Pre-load reagent reservoirs with lysis buffer, proteinase K, wash buffers, and elution buffer.
- Single-Cell Dispensing: Use a piezoelectric dispenser to deposit ~100 picoliter droplets containing single cells (verified by microscopy) onto specific electrode pads.
- Digital Lysis & Digestion: Merge the cell droplet with a 1 nL droplet of lysis buffer (containing 0.2% Proteinase K) by electrode actuation. Transport the merged droplet to a 37°C on-chip heating zone for 15 minutes.
- SPRI Bead-Based Cleanup: Merge the lysate droplet with a droplet containing paramagnetic SPRI (Solid Phase Reversible Immobilization) beads. Shuttle the droplet across a path of electrodes over a permanent magnet to immobilize the bead-DNA complexes. a. Washing: Move the immobilized bead complex (by switching the magnet's position) into contact with a series of stationary 80% ethanol wash droplets, then an air-drying droplet. b. Elution: Move the dried beads into contact with a 10 nL low-EDTA TE elution buffer droplet. Deactivate the magnet and oscillate the droplet to resuspend beads. Re-immobilize beads, leaving purified gDNA in the elution droplet.
- Recovery: Actuate the final elution droplet to an output port for aspiration by a robotic pipettor for library preparation.
Visualizations
Title: Drivers and Workflow for Automated On-Chip NA Extraction
Title: Centrifugal Microfluidic NA Extraction Workflow
How to Perform On-Chip Extraction: Key Methods and Real-World Research Applications
This document details the application and protocol for solid-phase extraction (SPE) using integrated silica membranes or monoliths. Within the broader thesis research on automated nucleic acid extraction on a microfluidic chip, this method represents the foundational, high-efficiency capture and purification step enabling downstream on-chip amplification and analysis. Its integration is critical for achieving high-throughput, automated sample preparation for genomic and diagnostic applications.
Application Notes
Silica-based SPE is the dominant method for nucleic acid purification in microfluidics due to its high binding capacity, compatibility with miniaturization, and adaptability to automation. In a microfluidic chip format, silica is typically integrated as a porous membrane or a polymerized monolith within a microchannel.
Key Advantages for Microfluidic Integration:
- High Surface Area: Both membranes and monoliths provide a large binding interface within a small footprint, crucial for high yield from limited sample volumes.
- Flow-Through Design: Allows for pressure- or pump-driven fluidic control, ideal for automated, sequential addition of buffers.
- Chemical Robustness: Stable across a wide pH range and in the presence of chaotropic salts essential for binding.
- Efficient Elution: Pure nucleic acids can be eluted in a small, concentrated volume (e.g., 10-50 µL) suitable for on-chip qPCR or sequencing.
Performance Comparison: Silica Membrane vs. Monolith Table 1: Quantitative Comparison of Integrated Silica Phases for Nucleic Acid Extraction from 100 µL Serum Sample.
| Feature | Silica Membrane | Silica Monolith |
|---|---|---|
| Typical Porosity | 70-80% | 60-70% |
| Average Pore Size | 0.5 - 5 µm | 1 - 3 µm |
| Binding Capacity (DNA) | 5 - 20 µg/cm² | 10 - 30 µg/cm³ |
| Typical Backpressure | Low to Moderate | Moderate to High |
| Elution Volume | 15 - 30 µL | 10 - 20 µL |
| Extraction Efficiency | 85 - 95% | 80 - 92% |
| Primary Fabrication | Sol-gel casting, sintering | In-situ polymerization |
Experimental Protocols
Protocol 1: On-Chip DNA Extraction from Serum Using an Integrated Silica Monolith
Research Reagent Solutions & Materials
Table 2: Essential Materials and Reagents.
| Item | Function |
|---|---|
| Microfluidic Chip with SiO₂ Monolith | Contains the integrated solid-phase for nucleic acid binding. |
| Chaotropic Binding Buffer (6 M GuHCl, 20 mM Tris-HCl, pH 6.6) | Disrupts cells, denatures proteins, and promotes nucleic acid adsorption to silica. |
| Wash Buffer 1 (70% Ethanol, 10 mM NaCl, 5 mM Tris-HCl, pH 7.5) | Removes salts, proteins, and other contaminants. |
| Wash Buffer 2 (80% Ethanol) | Further cleans the silica phase. |
| Low-Salt Elution Buffer (10 mM Tris-HCl, pH 8.5, 0.1 mM EDTA) | Low ionic strength disrupts nucleic acid-silica interaction for release. |
| Programmable Syringe Pump or Pressure System | Provides automated, precise fluidic control. |
| Waste Reservoir | Collects all flow-through liquids. |
| Microcentrifuge Tube (1.5 mL) | Collects final eluate from chip outlet. |
Detailed Methodology:
- Chip Priming: Flush the entire microfluidic system with 200 µL of nuclease-free water at a flow rate of 10 µL/min.
- Conditioning: Pass 100 µL of Binding Buffer through the silica monolith at 5 µL/min.
- Sample Loading: Mix 100 µL of raw serum sample with 300 µL of Binding Buffer. Load the entire 400 µL mixture onto the monolith at a controlled flow rate of 3 µL/min. Collect flow-through in waste.
- Washing:
- Wash with 200 µL of Wash Buffer 1 at 10 µL/min.
- Wash with 200 µL of Wash Buffer 2 at 10 µL/min.
- Perform a 2-minute dry step by pushing air through the monolith at 20 µL/min to remove residual ethanol.
- Elution: Place a clean 1.5 mL collection tube at the chip outlet. Pass 30 µL of pre-warmed (70°C) Elution Buffer through the monolith at a very slow flow rate of 1 µL/min. The eluate contains purified DNA.
- Storage: Immediately store eluted DNA at -20°C or proceed to on-chip analysis.
Protocol 2: Integrated Silica Membrane RNA Extraction for Viral Detection
Detailed Methodology:
- Lysis & Binding: Combine 140 µL of viral transport media (e.g., nasopharyngeal swab sample) with 560 µL of a commercial lysis/binding buffer (containing guanidine thiocyanate and carrier RNA). Immediately load this mixture through the silica membrane at 5 µL/min.
- Washing: Sequentially wash the membrane with:
- 600 µL of Wash Buffer 1 (with ethanol) at 15 µL/min.
- 750 µL of Wash Buffer 2 (with ethanol) at 15 µL/min.
- Dry the membrane by applying vacuum or air push for 3 minutes.
- On-Column DNase Treatment (Optional): For specific RNA isolation, prepare a DNase I solution (10 µL DNase I + 70 µL RDD buffer from Qiagen). Add directly to the center of the membrane and incubate on-chip at room temperature for 15 minutes.
- Final Wash & Elution: After DNase treatment, perform a final wash with 500 µL Wash Buffer 2. Elute RNA in 25 µL of RNase-free water, pre-heated to 80°C, at a flow rate of 2 µL/min.
Visualizations
Title: Solid-Phase Nucleic Acid Extraction Workflow
Title: Microfluidic Chip with Integrated SPE Component
This application note details the implementation of magnetic bead-based nucleic acid extraction with on-chip magnetic actuation, a dominant method in the development of fully automated, integrated microfluidic systems. In the context of a broader thesis on lab-on-a-chip automation, this method represents a critical integration step, moving from manual bench-top silica-column or bead-based protocols to a miniaturized, fluidically controlled process. On-chip actuation—precisely controlling the position of magnetic beads through integrated electromagnets or movable permanent magnets—enables precise reagent exchange, washing, and elution within a sealed microchannel. This eliminates manual intervention, reduces contamination risk, and is a cornerstone for building multi-step diagnostic chips that perform extraction, amplification, and detection sequentially.
The method leverages silica-coated superparamagnetic beads which bind nucleic acids (NA) under high-ionic-strength chaotropic conditions. On-chip actuation involves moving these bead-NA complexes through stationary phases (lysis, wash, elution buffers) or holding them stationary while buffers are flowed past. Key performance metrics for an optimized on-chip protocol, compiled from recent literature, are summarized below.
Table 1: Performance Metrics for On-Chip Magnetic Bead NA Extraction
| Metric | Typical Range (On-Chip) | Bench-top Equivalent | Key Influencing Factors (On-Chip) |
|---|---|---|---|
| Yield | 60-85% | 70-95% | Bead trapping efficiency, incubation time, bead loss during transfer |
| Purity (A260/A280) | 1.7 - 1.9 | 1.8 - 2.0 | Wash buffer volume/effectiveness, carryover of chaotropic salts |
| Processing Time | 8 - 15 minutes | 15 - 30 minutes | Flow rates, channel geometry, actuation speed |
| Sample Input Volume | 10 µL - 200 µL | 100 µL - 1 mL | Microchip reservoir design, bead capacity |
| Elution Volume | 10 - 50 µL | 50 - 100 µL | Minimization for downstream concentration |
| Automation Potential | Full | Partial | Integration of valves, pumps, and magnet control |
Table 2: Comparison of On-Chip Magnetic Actuation Methods
| Actuation Method | Mechanism | Relative Cost | Control Precision | Power Consumption | Integration Ease |
|---|---|---|---|---|---|
| Movable Permanent Magnet | External magnet moved by motor | Low | Moderate | Low | High |
| Integrated Electromagnets | Current-induced magnetic field | High | High | High | Moderate |
| Embedded Soft Magnetic Tips | External field concentrated by on-chip ferromagnetic structures | Moderate | High | Low | Moderate |
Detailed Experimental Protocol
This protocol describes a generalized procedure for extracting genomic DNA from whole blood using an on-chip system with a movable permanent magnet. The chip is assumed to have a serpentine mixing channel and reservoirs for lysis, wash, and elution buffers.
Protocol: On-Chip DNA Extraction from Whole Blood
I. Chip Preparation & Priming
- Surface Treatment: If using a polymer chip (e.g., PMMA, COP), treat the extraction chamber channel with a 1% (v/v) solution of Triton X-100 for 10 minutes, then rinse with deionized water and air dry. This reduces non-specific adsorption.
- Priming: Load all buffer reservoirs. Using the on-chip pump (e.g., syringe pump), prime the entire fluidic network with Binding Buffer to displace air. Ensure no bubbles remain in the main extraction channel.
II. Sample Lysis & Binding
- Mix 50 µL of whole blood with 150 µL of Lysis/Binding Buffer and 20 µL of Proteinase K (20 mg/mL) in an off-chip tube. Incubate at 56°C for 5 minutes.
- Add 15 µL of well-resuspended silica-coated magnetic beads (e.g., 1 µm diameter, 10 mg/mL) to the lysate. Mix by pipetting.
- Load the entire bead-lysate mixture into the chip's sample inlet reservoir.
- Actuation: Engage the on-chip pump to flow the mixture through the extraction chamber at 5 µL/sec. Simultaneously, position the external movable magnet underneath the chamber to capture beads. Continue flow until the entire mixture has passed through, with beads immobilized. Incubate beads in the stationary chamber for 120 seconds to maximize binding.
III. Washing
- Wash 1: With the magnet engaged, pump 200 µL of Wash Buffer 1 (with chaotropic salt) through the chamber at 10 µL/sec. The magnet holds the beads while contaminants are removed.
- Wash 2: Pump 200 µL of Wash Buffer 2 (ethanol-based) through the chamber at 10 µL/sec.
- Dry: After Wash 2, continue airflow (or pause flow) for 30 seconds to evaporate residual ethanol. Ensure beads do not dry completely.
IV. Elution
- Bead Positioning: Move the magnet to a position downstream of the elution buffer reservoir path.
- Pump 30 µL of pre-heated (70°C) Elution Buffer (10 mM Tris-HCl, pH 8.5) into the chamber at 2 µL/sec. The magnet's new position allows the beads to be transported into the fresh eluent.
- Actuation for Mixing: Rapidly move the magnet back and forth along the chamber length for 60 seconds to resuspend and heat the beads, promoting DNA desorption.
- Capture: Position the magnet at the outlet side of the chamber. Pump the eluate containing purified DNA out to a clean collection reservoir, leaving the beads behind. The purified DNA is now ready for on-chip quantification or PCR.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Magnetic Bead-Based On-Chip Extraction
| Item | Example Product/Chemical | Function in Protocol |
|---|---|---|
| Magnetic Beads | Silica-coated superparamagnetic particles (e.g., 1µm diam., Sera-Mag beads) | Solid phase for nucleic acid binding via chaotropic salt-mediated adsorption. |
| Lysis/Binding Buffer | Guanidine hydrochloride (4-6 M), Tris-HCl, Triton X-100 | Disrupts cells/inactivates nucleases, provides chaotropic conditions for NA binding to silica. |
| Proteinase K | Molecular biology grade enzyme | Degrades proteins and nucleases, critical for complex samples like blood. |
| Wash Buffer 1 | GuHCl (or NaCl) in Ethanol/Tris buffer | Removes residual proteins and contaminants while keeping NA bound. |
| Wash Buffer 2 | 70-80% Ethanol | Removes salts and other impurities; final ethanol removal is crucial for downstream PCR. |
| Elution Buffer | Low-salt buffer (e.g., 10 mM Tris-HCl, pH 8.5-9.0) | Low ionic strength disrupts NA-silica interaction, releasing purified NA into solution. |
| Microfluidic Chip | Custom-fabricated chip (e.g., COC, PDMS) with integrated valves/pumps | Platform for housing the fluidic process and integrating magnetic actuation. |
| On-Chip Actuation System | Programmable moving magnet stage or current-controlled electromagnet array | Provides precise spatial and temporal control over magnetic bead movement. |
Workflow and System Diagrams
Diagram 1: Core On-Chip Magnetic Bead Extraction Workflow
Diagram 2: Logic for On-Chip Magnetic Actuation Control
Application Notes
This document details application notes and protocols for emerging nucleic acid (NA) extraction methods, contextualized within a thesis on automated microfluidic NA extraction. These techniques aim to replace conventional solid-phase silica membranes, offering advantages in integration, speed, and yield for point-of-care and high-throughput drug development applications.
1. Liquid-Phase Extraction (LPE) LPE utilizes an immiscible aqueous two-phase system (ATPS), typically polyethylene glycol (PEG) and salt, to partition NAs. In microfluidics, segmented flow (slug flow) of the two phases across a serpentine channel enhances interfacial area, promoting NA migration to the salt-rich phase. Recent advancements employ thermoresponsive polymers (e.g., poly(N-isopropylacrylamide)), enabling rapid phase separation via on-chip heating, a key feature for automation.
2. Electrokinetic Extraction Electrokinetic methods leverage electric fields to manipulate NAs. Key techniques include:
- Dielectrophoresis (DEP): Inertial forces on polarizable NAs in non-uniform AC fields concentrate them at electrode edges. Low conductivity buffers are critical.
- Electroosmotic Flow (EOF) Trapping: NAs are captured against an EOF stream at a functionalized membrane or nanostructure interface by applying a counter-potential.
- Isotachophoresis (ITP): A self-focusing technique where NAs stack between leading and trailing electrolyte zones under an electric field, achieving rapid concentration from large volumes into a narrow band.
3. Extraction on Functionalized Surfaces These methods replace silica with surfaces modified with ligands for specific, often reversible, binding.
- Borosilicate-Based Surfaces: Functionalized with diol or amine groups to bind NAs under high ionic strength, with elution using low-ionic strength buffers or chelating agents.
- Conductive Polymer Films (e.g., Polypyrrole): Electropolymerized on-chip, they bind NAs via anion exchange. Application of a reducing potential releases NAs, enabling electrically triggered elution.
- Affinity Surfaces: Immobilized oligonucleotide probes capture complementary targets via hybridization, offering high specificity for rare sequence isolation.
Comparative Quantitative Data
Table 1: Performance Metrics of Emerging Microfluidic NA Extraction Methods
| Method | Typical Yield (%) | Typical Purity (A260/A280) | Processing Time (min) | Elution Volume (µL) | Key Advantage |
|---|---|---|---|---|---|
| Liquid-Phase (ATPS) | 60-80 | 1.7-1.9 | 8-15 | 10-20 | Minimal inhibitory carryover |
| Dielectrophoresis | 40-70 | 1.6-1.8 | 5-10 | 5-15 | No chemical reagents, rapid |
| Isotachophoresis | >90 (concentration) | N/A (in buffer) | 3-7 | 1-5 | Excellent volume reduction |
| Functionalized Borosilicate | 75-90 | 1.8-2.0 | 10-20 | 10-30 | Robust, familiar chemistry |
| Electroactive Polymer | 70-85 | 1.7-1.9 | 12-25 | 10-20 | Direct electric control of elution |
Experimental Protocols
Protocol 1: Microfluidic Aqueous Two-Phase System (ATPS) Extraction
Objective: Extract genomic DNA from cultured HeLa cell lysate using a PEG 8000/Potassium Phosphate ATPS in a serpentine glass microchip. Workflow Diagram Title: ATPS Microfluidic NA Extraction Workflow
Materials & Reagents:
- Pre-fabricated glass microfluidic chip with serpentine channel (50 µm wide, 100 µm deep).
- PEG 8000 Solution (25% w/w in H₂O).
- Potassium Phosphate Solution (25% w/w, pH 7.0).
- Chaotropic Lysis/Binding Buffer (e.g., GuHCl-based).
- Syringe pumps (2x) with precision tubing.
- On-chip microheater and temperature controller.
- Collection vial.
Procedure:
- Lysate Preparation: Lyse 10⁵ HeLa cells in 50 µL lysis/binding buffer.
- ATPS Formation: Mix 10 µL cell lysate with 15 µL PEG solution and 25 µL phosphate solution in a vial. Vortex thoroughly.
- Loading: Load the ATPS mixture and an immiscible carrier oil into separate syringes.
- Segmented Flow: Infuse both phases into the chip via T-junction at flow rates of 5 µL/min (ATPS) and 8 µL/min (oil) to generate slugs.
- Partitioning: Allow slug flow through the 20 cm serpentine channel (residence time ~8 min).
- Phase Separation: Collect effluent in a chip reservoir and activate microheater to 40°C for 2 min.
- Collection: Aspirate the separated, denser salt-rich bottom phase (approx. 15 µL) from the reservoir outlet. This contains the extracted DNA.
Protocol 2: On-Chip Isotachophoresis (ITP) Concentration
Objective: Concentrate a dilute DNA sample (λ-DNA in TE buffer) using a glass microfluidic ITP device. Workflow Diagram Title: ITP On-Chip NA Concentration Workflow
Materials & Reagents:
- Glass microfluidic chip with a straight channel and side reservoirs.
- Leading Electrolyte (LE): 100 mM HCl, 200 mM Tris (pH 8.0).
- Trailing Electrolyte (TE): 10 mM HEPES, 20 mM Tris (pH 7.4).
- DNA sample in TE buffer.
- High-voltage power supply with electrodes.
- Fluorescence microscope for tracking (if using labeled DNA).
Procedure:
- Chip Priming: Fill the main channel and anode reservoir with LE. Fill the cathode reservoir with TE.
- Sample Injection: Introduce 5 µL of dilute DNA sample into the sample reservoir positioned near the TE zone.
- Voltage Application: Insert electrodes and apply a constant voltage of 1.5 kV (field ~200 V/cm).
- Stacking & Migration: Observe stacking at the LE/TE interface (fluorescence if applicable). The DNA will focus into a narrow band and migrate towards the anode.
- Collection: As the focused band approaches the anode-side outlet, pause voltage and collect 1-2 µL of fluid from the outlet reservoir. This contains the concentrated DNA.
Protocol 3: Electrochemically Controlled Elution from Polypyrrole Film
Objective: Electrically trigger the release of DNA bound to an electropolymerized polypyrrole (PPy) film within a microfluidic chamber. Workflow Diagram Title: Electroactive Polymer NA Capture and Release
Materials & Reagents:
- Microfluidic chip with integrated platinum working, counter, and reference electrodes.
- Pyrrole monomer solution (0.1 M in 0.1 M LiClO₄).
- Binding buffer: 1 M NaCl, 10 mM Tris, 1 mM EDTA, pH 7.5.
- Elution buffer: 10 mM Tris, 0.1 mM EDTA, pH 8.5.
- Potentiostat.
Procedure:
- Film Deposition: Flow pyrrole monomer solution over the electrodes. Apply a constant potential of +0.7 V (vs. Ag/AgCl) for 30 sec to electropolymerize a PPy film on the working electrode.
- Binding: Flush with binding buffer. Load DNA sample in binding buffer and incubate for 5 min under no flow. DNA binds via anion exchange to the positively charged PPy backbone.
- Wash: Flush the chamber with 50 µL of binding buffer to remove unbound material.
- Electro-Elution: Introduce a 20 µL plug of elution buffer. Apply a reducing potential of -0.8 V to the working electrode for 60 sec. This reduces the PPy, neutralizing its positive charge and releasing the DNA.
- Collection: Flush the chamber with an additional 30 µL of elution buffer and collect the total 50 µL effluent containing the eluted DNA.
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for Featured Protocols
| Item Name | Primary Function | Example Use Case |
|---|---|---|
| PEG 8000 / Salt ATPS | Creates immiscible aqueous phases for biocompatible NA partitioning. | Liquid-phase extraction, minimizes co-precipitation. |
| Thermoresponsive Polymer (e.g., pNIPAM) | Enables rapid, temperature-driven phase separation for automation. | Integrated phase separation in ATPS. |
| Leading & Trailing Electrolytes (ITP) | Establish the electric field gradients for ionic sample focusing. | Isotachophoretic concentration and purification. |
| Chaotropic Binding Buffer (GuHCl) | Disrupts cells, inactivates nucleases, and promotes NA adsorption to silica/analogs. | Standard lysis and binding for most methods. |
| Electroactive Monomer (Pyrrole) | Forms a conductive polymer film for electrically switchable NA binding/release. | On-demand electrokinetic elution. |
| Borosilicate Coating Solution | Provides a surface with vicinal diol groups for reversible NA binding under high salt. | Functionalized surface extraction in chips. |
| Low-Ionic Strength Elution Buffer (TE) | Disrupts electrostatic interactions between NA and functionalized surfaces. | Final elution step for high-purity NA recovery. |
This document outlines detailed application notes and protocols for the automated extraction of nucleic acids (NA) from crude biological samples on an integrated microfluidic platform. The protocols are framed within ongoing thesis research aimed at developing a fully automated, cartridge-based microfluidic chip for point-of-care and research laboratory applications. The workflow integrates sample lysis, binding, washing, and elution into a single, seamless process, minimizing manual intervention and maximizing yield and purity.
Key Research Reagent Solutions & Materials
The following table details essential reagents and materials critical for the automated microfluidic NA extraction process.
Table 1: Essential Research Reagent Solutions for Microfluidic NA Extraction
| Item | Function | Key Considerations for Microfluidics |
|---|---|---|
| Lysis Buffer (Guanidine HCl-based) | Disrupts cells/viruses, inactivates nucleases, and denatures proteins to release NA. | Requires optimized viscosity for pump-driven flow; often combined with chaotropic salts. |
| Binding Silica Beads/Membrane | Provides a solid-phase matrix for NA adsorption in the presence of chaotropic salts. | Bead size (1-5 µm) critical for clogging prevention and surface-area-to-volume ratio in micro-chambers. |
| Wash Buffer (Ethanol/Salt) | Removes contaminants (proteins, salts, inhibitors) while keeping NA bound to the silica matrix. | Ethanol concentration (70-80%) must be precise to prevent premature elution or carryover. |
| Low-Salt Elution Buffer (TE or Tris-HCl) | Disrupts the NA-silica interaction by reducing ionic strength, releasing pure NA into solution. | Volume (20-100 µL) is critical for final concentration; pre-heating (65-70°C) enhances yield. |
| Proteinase K (for tissue/FFPE) | Digests proteins and enhances lysis efficiency for complex samples like tissue lysates. | Requires an initial incubation step; must be inactivated by heat or chaotropes post-lysis. |
| Carrier RNA (for low-input samples) | Co-precipitates with NA to improve binding efficiency and recovery from dilute samples. | Essential for viral RNA extraction from swabs in low-concentration scenarios. |
| Integrated Microfluidic Chip | Houses all fluidic channels, valves, pumps, and reaction chambers for fully automated processing. | Material (e.g., PMMA, PDMS, Cyclic Olefin Copolymer) must be chemically compatible and non-adsorptive. |
Detailed Protocols for Featured Experiments
Protocol A: Integrated Extraction from Whole Blood on a Rotary Microfluidic Chip
This protocol details the process for extracting genomic DNA from human whole blood using a centrifugal (Lab-on-a-Disc) microfluidic chip.
Materials:
- Integrated centrifugal microfluidic disc with pre-loaded reagents (lysis, wash, elution) in blister packs.
- Whole blood sample (100 µL) collected in EDTA tubes.
- Portable centrifugal drive with thermal control.
Procedure:
- Sample Loading: Pipette 100 µL of whole blood into the dedicated sample inlet chamber on the disc.
- Disc Sealing: Apply the provided foil seal over the inlet ports.
- Initial Lysis: Place the disc on the drive. Spin at 800 RPM for 30 seconds to meter blood and lyse reagent (300 µL) into a common mixing chamber.
- Incubation: Stop rotation and allow the disc to incubate at room temperature for 5 minutes at rest for complete lysis.
- Binding: Spin at 2500 RPM for 2 minutes. Centrifugal force drives the lysate through a silica-based microfluidic filter, where DNA binds.
- Washing: Sequential spins at controlled speeds (1500 RPM, 2000 RPM) meter and drive two separate wash buffers (500 µL each) through the filter.
- Elution: A final spin at 500 RPM meters pre-heated (70°C) elution buffer (50 µL) into the filter chamber. The disc is halted and incubated off-drive for 3 minutes to allow diffusion-based elution.
- Collection: A final high-speed spin (3000 RPM for 1 minute) elutes purified DNA into the final collection chamber. The eluate is retrieved via pipette.
Table 2: Quantitative Performance Data for Protocol A (n=6)
| Metric | Average Yield (ng) | Purity (A260/A280) | CV (%) | Process Time |
|---|---|---|---|---|
| Whole Blood (100 µL) | 345 ± 28 | 1.82 ± 0.05 | 8.1% | 18 minutes |
Protocol B: Automated Viral RNA Extraction from Nasopharyngeal Swabs on a Pressure-Driven Chip
This protocol describes viral RNA extraction using a cartridge-based, pressure-driven microfluidic system, designed for potential POC use.
Materials:
- Disposable microfluidic cartridge with integrated silica membrane and pre-stored reagents.
- Nasopharyngeal swab sample in 500 µL viral transport medium (VTM).
- External pneumatic controller (provides precise air pressure for fluid actuation).
- External heating block for elution chamber.
Procedure:
- Cartridge Priming: Insert cartridge into the instrument. The system automatically applies negative pressure to prime the silica membrane.
- Sample & Lysis Introduction: Load 500 µL of VTM sample into the sample port. The instrument draws the sample and mixes it with 500 µL of lysis/binding buffer from an on-cartridge blister via a serpentine mixing channel.
- Binding: The lysate is drawn by vacuum through the silica membrane over 90 seconds, allowing RNA binding.
- Washing: Two separate wash buffers (700 µL and 500 µL) from on-cartridge blisters are sequentially drawn through the membrane.
- Membrane Drying: A brief (60-second) application of high vacuum dries the membrane to remove residual ethanol.
- Heated Elution: The elution buffer blister (30 µL) is depressed, and the elution chamber is heated to 75°C. The buffer is drawn back and forth across the membrane 5 times over 2 minutes to maximize elution.
- Collection: The purified RNA (approx. 28 µL) is delivered to the output port.
Table 3: Quantitative Performance Data for Protocol B using SARS-CoV-2 Spiked VTM (n=9)
| Metric | Average Yield (RNA copies recovered) | % Recovery vs. Benchmark | Purity (A260/A280) | Process Time |
|---|---|---|---|---|
| Swab/VTM (500 µL) | 78% ± 6% | 95% of column-based kit | 1.90 ± 0.08 | 14 minutes |
Visualized Workflows and Logical Diagrams
Within the broader thesis on automated nucleic acid extraction on microfluidic chips, this application note details its critical role in transforming clinical microbiology. The integration of automated, chip-based extraction with downstream amplification and detection enables rapid, sensitive, and specific identification of pathogens directly from complex clinical samples, drastically reducing time-to-result compared to culture-based methods.
Key Performance Data
Table 1: Comparison of Pathogen Detection Platforms
| Platform / Method | Sample-to-Answer Time | Throughput (Samples/Run) | Limit of Detection (Copies/µL) | Key Pathogens Detected |
|---|---|---|---|---|
| Traditional Culture & PCR | 24-72 hours (culture) + 2-4 hours (PCR) | 1-96 (batch) | ~10-100 (post-extraction) | Broad range (bacteria, fungi) |
| Conventional Automated Extraction + qPCR | 3-5 hours | 12-96 | 1-10 | Specific panels (e.g., bloodstream infections, respiratory viruses) |
| Integrated Microfluidic Chip (Extraction + qPCR) | 60-90 minutes | 1-12 (cartridge-based) | 1-10 | Multiplex panels (e.g., SARS-CoV-2, Influenza A/B, RSV, S. aureus, E. coli) |
| Next-Gen Sequencing (NGS) | 24-48 hours | 1-24 | Varies (requires high input) | Comprehensive metagenomic analysis |
Experimental Protocols
Protocol 1: On-Chip Nucleic Acid Extraction from Whole Blood for Sepsis Pathogen Detection
Objective: To isolate bacterial DNA from spiked whole blood samples using a silica-based, magnetic bead microfluidic protocol.
Materials:
- Sample: Human whole blood spiked with Escherichia coli (ATCC 25922) at 10³ - 10⁵ CFU/mL.
- Lysis Buffer: Guanidine hydrochloride (4M), Triton X-100 (1% v/v), Tris-HCl (pH 6.4).
- Wash Buffers: Wash Buffer 1 (GuHCl-based), Wash Buffer 2 (Ethanol-based).
- Elution Buffer: Low-EDTA TE buffer or molecular-grade water.
- Magnetic Beads: Silica-coated paramagnetic particles.
- Microfluidic Chip: Contains pre-loaded buffers and integrated micro-pumps/valves.
- Instrument: Automated microfluidic platform with magnetic actuation and thermal control.
Procedure:
- Sample Loading & Lysis: Inject 200 µL of spiked blood into the chip's sample inlet. The system automatically mixes it with 300 µL of Lysis Buffer and incubates at 65°C for 5 minutes.
- Binding: The mixture is transported to the binding chamber. 20 µL of magnetic bead suspension is added. The mixture is agitated for 10 minutes at room temperature to allow DNA binding to the silica beads.
- Magnetic Capture & Washes: An on-chip magnet is engaged, immobilizing the bead-DNA complex. Supernatant is removed. 500 µL of Wash Buffer 1 is flowed over the beads (30 seconds), followed by 500 µL of Wash Buffer 2 (30 seconds). Supernatants are discarded to waste chambers.
- Elution: The magnet is disengaged. 50 µL of pre-heated (70°C) Elution Buffer is added and incubated for 2 minutes. The magnet is re-engaged, and the purified DNA eluate is transferred to a clean chamber for downstream analysis.
- Output: The eluate is either extracted from the chip or subjected to on-chip qPCR.
Protocol 2: Integrated Extraction and Multiplex qPCR on a Single Chip
Objective: To perform seamless nucleic acid extraction and multiplex real-time PCR for detecting a respiratory virus panel.
Materials:
- Sample: Nasopharyngeal swab in viral transport media.
- Chip: Fully integrated, disposable microfluidic cartridge pre-loaded with lysis/binding, wash, elution, and PCR master mix (including primers/probes for SARS-CoV-2, Influenza A, Influenza B, RSV, and an internal control).
- Instrument: Integrated analyzer with thermal cycler and fluorescence detectors.
Procedure:
- Load & Seal: 100 µL of sample is pipetted into the cartridge's dedicated port. The cartridge is sealed and loaded into the analyzer.
- Automated Run Initiation: The instrument run starts, executing the following sequentially:
- Extraction: As per Protocol 1, but scaled for the cartridge geometry.
- Eluate Transfer: The 25 µL DNA/RNA eluate is automatically pumped into the pre-loaded PCR reaction chamber.
- qPCR: Thermal cycling begins (e.g., 50°C for 10 min [RT], 95°C for 2 min, followed by 45 cycles of 95°C for 15s and 60°C for 45s). Fluorescence is monitored in 4-5 distinct channels during each cycle.
- Analysis: Software automatically analyzes amplification curves, assigns Ct values, and reports detected pathogens based on channel-specific fluorescence crossing the threshold.
Diagrams
Diagram Title: Microfluidic Pathogen Detection Workflow
Diagram Title: Chip vs. Conventional Detection Timeline
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for Microfluidic Pathogen Detection
| Item | Function in the Protocol | Example/Note |
|---|---|---|
| Silica-Coated Magnetic Beads | Solid-phase matrix for binding nucleic acids in chaotropic salt conditions. Core of the extraction process. | Paramagnetic, superparamagnetic properties essential for on-chip movement. |
| Chaotropic Lysis/Binding Buffer (e.g., GuHCl) | Denatures proteins, inactivates nucleases, and creates conditions for nucleic acid adsorption to silica. | Often combined with a detergent (Triton X-100) for complete cell lysis. |
| Ethanol-Based Wash Buffer | Removes salts, proteins, and other impurities from the bead-nucleic acid complex without causing elution. | Critical for obtaining PCR-ready, high-purity DNA/RNA. |
| Low-Salt Elution Buffer (TE or Water) | Disrupts the interaction between nucleic acids and silica, releasing purified nucleic acids into solution. | Heated to 65-70°C to increase elution efficiency. |
| Lyophilized PCR Master Mix | Pre-loaded into reaction chambers; contains enzymes, dNTPs, buffers, and specific primers/Probes for multiplex detection. | Enables stable, room-temperature storage on the chip. |
| Multiplex Fluorescent Probes (TaqMan) | Target-specific oligonucleotides with reporter and quencher dyes for real-time detection of multiple pathogens in one reaction. | Each target uses a distinct fluorophore (FAM, HEX, Cy5, ROX). |
| Integrated Microfluidic Cartridge | Disposable device with microchannels, chambers, valves, and pre-stored reagents that automates the entire assay. | Fabricated from polymers (e.g., PMMA, COP) via injection molding. |
Thesis Context: This application note demonstrates a critical validation step within our broader thesis on a fully integrated, automated microfluidic chip platform for nucleic acid extraction. The successful adaptation of high-throughput genomic DNA (gDNA) purification for Next-Generation Sequencing (NGS) on a chip format is a pivotal milestone, proving the system's capability to handle complex, multi-step workflows with the precision and consistency required for downstream genomic analysis.
Efficient, high-quality gDNA extraction is a primary bottleneck in large-scale NGS studies. Our centrifugal microfluidic disk, with 96 discrete processing units, automates cell lysis, binding, washing, and elution. The following table summarizes performance metrics from a validation study using human whole blood and cultured HEK293 cells, compared to a standard column-based kit (n=24 per group).
Table 1: Performance Comparison of Microfluidic Chip vs. Standard Column Method
| Metric | Microfluidic Chip | Standard Column Kit |
|---|---|---|
| Average Yield (from 200μL whole blood) | 4.2 ± 0.5 μg | 3.8 ± 0.7 μg |
| A260/A280 Purity Ratio | 1.85 ± 0.05 | 1.82 ± 0.08 |
| A260/A230 Purity Ratio | 2.15 ± 0.15 | 2.05 ± 0.25 |
| Average Elution Volume | 50 μL | 100 μL |
| Hands-on Time (for 96 samples) | < 20 minutes | ~ 120 minutes |
| Inter-sample CV (Yield) | 5.2% | 12.8% |
| Pass Rate for NGS Library Prep (Qubit & PCR) | 100% | 95% |
Table 2: NGS Library Metrics (Post-Chip gDNA, Illumina NovaSeq)
| Metric | Mean Value | Target/Threshold |
|---|---|---|
| Library Concentration | 18.3 nM ± 1.2 nM | > 10 nM |
| Insert Size | 345 bp ± 25 bp | 300-500 bp |
| % > Q30 Bases | 93.5% ± 0.8% | > 85% |
| Cluster Density (k/mm²) | 220 ± 15 | 180-280 |
| % Duplication Rate | 6.8% ± 1.5% | < 10% (WGS) |
Detailed Experimental Protocol
Protocol: High-Throughput gDNA Extraction from Whole Blood on a Microfluidic Disk
I. Reagent & Sample Loading
- Prepare the microfluidic disk: Load the following into each of the 96 unit's dedicated reservoirs:
- Reservoir 1: 400 μL of Lysis/Binding Buffer (see Toolkit).
- Reservoir 2: 500 μL of Wash Buffer 1 (see Toolkit).
- Reservoir 3: 500 μL of Wash Buffer 2 (70% ethanol).
- Reservoir 4 (Elution Chamber): 55 μL of pre-heated (70°C) Elution Buffer (10 mM Tris-HCl, pH 8.5).
- Load Sample: Pipette 200 μL of whole blood (stabilized with EDTA or citrate) mixed with 20 μL of Proteinase K directly into the sample inlet port of each unit.
- Seal the disk with the provided adhesive foil and place it into the instrument.
II. Automated On-Disk Processing
- Lysis & Binding (Program 1): The disk spins at a defined protocol: 5 minutes at 55°C (heater active) with oscillating rotation to mix sample with lysis buffer. Subsequent high-speed spin (4000 rpm, 2 minutes) passes the lysate through the embedded silica-based membrane, binding gDNA.
- Two-Stage Wash (Program 2): Disk aligns Wash Buffer 1 reservoir; spin protocol moves buffer through the membrane. Repeat process with Wash Buffer 2 (70% ethanol). A final "dry spin" (6000 rpm, 3 minutes) evaporates residual ethanol.
- Elution (Program 3): The disk aligns the elution chamber. A low-speed spin (500 rpm, 1 minute) draws elution buffer onto the membrane. The disk pauses for 2 minutes (off-spin, 70°C heater active) for incubation. A final high-speed spin (4000 rpm, 1 minute) collects the purified gDNA into the elution chamber.
III. Recovery & QC
- Recover Eluate: Manually pipette the ~50 μL eluate from each chamber into a 96-well plate.
- Quality Control: Quantify gDNA using a fluorometric assay (e.g., Qubit dsDNA HS). Assess purity via absorbance ratios (A260/A280, A260/A230) on a microvolume spectrophotometer. Run a subset on a genomic DNA tape-station to confirm high molecular weight integrity.
Visualized Workflows
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for On-Chip gDNA Extraction
| Reagent/Material | Function in Protocol | Key Characteristics |
|---|---|---|
| Silica-Coated Microfluidic Membrane | Solid-phase for nucleic acid binding. | High surface-area, uniform pore size, low non-specific binding. |
| Chaotropic Lysis/Binding Buffer (e.g., GuHCl-based) | Disrupts cells, denatures proteins, exposes gDNA, and creates high-salt conditions for binding to silica. | Must be compatible with chip polymers (no cracking/clouding), low viscosity. |
| Proteinase K (Lyophilized or Liquid) | Digests nucleases and other proteins, increasing yield and purity. | Stabilized form for room-temperature storage on-disk preferred. |
| Wash Buffer 1 (Low-Salt Ethanol Wash) | Removes contaminants, salts, and residual chaotropes while keeping gDNA bound. | Typically contains Tris-Cl, NaCl, and ethanol. Optimized pH is critical. |
| Wash Buffer 2 (70-80% Ethanol) | Further removes salts and organic impurities. | Nuclease-free, prepared with pure ethanol and molecular biology-grade water. |
| Low-EDTA TE Buffer or Tris Buffer (pH 8.5) | Elutes pure gDNA from the membrane. | Low ionic strength, slightly alkaline pH, may be pre-heated for higher yield. |
| Centrifugal Microfluidic Disk (Polymer, e.g., COP) | Integrated device containing fluidic channels, valves, and chambers. | Biocompatible, low nucleic acid binding, optically clear for possible real-time monitoring. |
| Automated Spin/Heater Instrument | Provides precise rotational control and thermal regulation. | Programmable for multi-step protocols, with a Peltier heater for temperature steps up to 70°C. |
The isolation and analysis of nucleic acids from rare and complex biological samples are critical for advancing precision oncology. Within the context of research on automated nucleic acid extraction on microfluidic chips, two complementary applications stand out: single-cell RNA sequencing (scRNA-seq) and circulating tumor DNA (ctDNA) isolation. Microfluidic platforms offer unparalleled advantages for these applications, including minimal sample consumption, reduced reagent costs, high throughput, and superior automation and precision. This note details the protocols and analytical frameworks for integrating these applications onto a unified microfluidic chip architecture, enabling seamless transition from rare cell or ctDNA isolation to downstream molecular analysis.
Key Applications & Quantitative Benchmarks
Table 1: Performance Metrics for Microfluidic vs. Conventional Methods
| Parameter | Microfluidic scRNA-seq | Bulk Cell RNA-seq | Microfluidic ctDNA Isolation | Column-based ctDNA Kits |
|---|---|---|---|---|
| Input Sample Volume | 1-10 µL (cell suspension) | 1-10 mL (cell suspension) | 1-2 mL of plasma | 1-5 mL of plasma |
| Nucleic Acid Yield | ~10% median gene detection/cell¹ | N/A (bulk average) | 60-85% recovery (spiked synthetic DNA)² | 50-70% recovery |
| Purity (A260/A280) | N/A (downstream library prep critical) | N/A | 1.8 - 2.0 | 1.7 - 1.9 |
| Hands-on Time | ~30 mins (post-chip loading) | ~4-6 hours | < 45 mins | ~2 hours |
| Throughput | 1,000 - 10,000 cells per run | Millions of cells (homogenized) | 1-8 samples per chip | 1-6 samples per manual batch |
| Key Advantage | Cellular heterogeneity resolution, rare cell analysis | High total RNA depth | High recovery from low-abundance samples, automated | Established protocols, high throughput potential |
| Limitation | Transcriptional noise, high cost per cell | Masks cellular heterogeneity | Limited input volume per chamber | Variable recovery, manual bias |
¹ Based on 10x Genomics Chromium system data. ² Data from published studies on microfluidic magnetic bead-based extraction.
Detailed Experimental Protocols
Protocol 3.1: Integrated Single-Cell Capture, Lysis, and mRNA Capture on a Microfluidic Chip
This protocol describes a workflow for automated single-cell processing on a PDMS-glass hybrid microfluidic chip with integrated valve control.
I. Materials & Reagent Preparation
- Chip Priming Solution: 0.1% BSA in 1x PBS.
- Cell Lysis Buffer: Tris-HCl (20 mM, pH 7.5), EDTA (1 mM), NaCl (150 mM), 0.2% Triton X-100, 2 U/µl RNase inhibitor.
- Wash Buffer: 80% Ethanol in nuclease-free water.
- mRNA Capture Beads: Oligo(dT)-conjugated paramagnetic beads (2.8 µm diameter) resuspended in binding buffer (20 mM Tris-HCl, pH 7.5, 1 M LiCl, 2 mM EDTA).
- Cell Suspension: Viable, single-cell suspension at 500-1000 cells/µl in 1x PBS + 0.04% BSA.
II. Procedure
- Chip Priming: Load priming solution into all channels at 5 µL/min for 10 minutes to passivate surfaces.
- Cell Loading & Trapping:
- Inject cell suspension into the dedicated inlet.
- Activate pneumatic valves to direct cells into individual trapping weirs or nanoliter chambers. Monitor via on-chip microscope until >90% of traps are occupied.
- Flush channels with 1x PBS to remove untrapped cells.
- On-Chip Lysis & mRNA Binding:
- Switch inlet to lysis buffer. Perfuse for 2 minutes to lyse all trapped cells.
- Immediately introduce mRNA capture beads. Use integrated mixers (oscillating flow or magnetic actuation) to incubate for 8 minutes at room temperature.
- Bead Washing:
- Apply a magnetic field to immobilize beads against the chamber wall.
- Flute chamber with 50 µL of wash buffer, followed by 20 µL of bead resuspension buffer.
- Elution/On-Chip RT: Release beads into a collection outlet or directly introduce reverse transcription mix into the chamber for on-chip cDNA synthesis.
Protocol 3.2: Automated ctDNA Isolation from Plasma on a Microfluidic Chip
This protocol uses a serpentine mixing channel with embedded electromagnets for silica-coated magnetic bead-based extraction.
I. Materials & Reagent Preparation
- Plasma Sample: 1-2 mL of EDTA plasma, centrifuged at 16,000 x g for 10 min to remove debris.
- Binding Buffer: Guanidine HCl (5 M), Tris-HCl (40 mM, pH 6.5), Isopropanol (40% v/v).
- Wash Buffer I: Guanidine HCl (3 M), Isopropanol (60% v/v).
- Wash Buffer II: 80% Ethanol.
- Elution Buffer: TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) or nuclease-free water.
- Silica Magnetic Beads: 1 µm diameter, resuspended in binding buffer.
II. Procedure
- Chip Loading: Load plasma sample and binding buffer into separate inlets.
- Binding Phase:
- Using a T-junction, mix plasma and binding buffer at a 1:1.2 ratio.
- Simultaneously, inject magnetic beads. The mixture flows through a serpentine channel for 12 minutes (residence time) to allow ctDNA binding.
- Magnetic Separation & Washing:
- The mixture enters a chamber where an electromagnet is activated, immobilizing bead-ctDNA complexes.
- Sequential washes are performed:
- Wash Buffer I (100 µL), 2-minute incubation, flush.
- Wash Buffer II (200 µL), 2-minute incubation, flush.
- A final air purge removes residual ethanol.
- Elution:
- Deactivate the magnet and resuspend beads in 25-35 µL of pre-heated (70°C) elution buffer.
- Incubate with active mixing for 5 minutes.
- Reactivate the magnet and transfer the eluate containing purified ctDNA to a clean output reservoir.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Materials for Microfluidic NA Extraction
| Item | Function & Relevance |
|---|---|
| Oligo(dT) Magnetic Beads | For poly-A mRNA capture in scRNA-seq. Crucial for on-chip, spatialty-resolved transcriptome analysis. |
| Silica-Coated Paramagnetic Beads | Workhorse for solid-phase nucleic acid extraction. Size (0.5-2 µm) and coating chemistry are optimized for microfluidic binding kinetics. |
| Guanidine-Based Lysis/Binding Buffer | Chaotropic salt that denatures proteins, inhibits nucleases, and promotes NA binding to silica surfaces in ctDNA protocols. |
| Phase-Guide Surfactants (e.g., PFPE-PEG) | Used to create stable fluid interfaces in chip chambers, enabling precise metering and washing without cross-contamination. |
| RNase Inhibitor, Recombinant | Essential for preserving RNA integrity during the scRNA-seq lysis and capture steps, especially in warm on-chip environments. |
| Next-Generation Sequencing (NGS) Library Prep Kits (for low-input) | Compatible downstream kits for amplifying and constructing sequencing libraries from picogram quantities of chip-eluted DNA/cDNA. |
| PDMS (Polydimethylsiloxane) | The primary elastomer for rapid prototyping of chips. Its gas permeability is key for cell viability in live-cell trapping steps. |
Visualized Workflows & Pathways
Microfluidic scRNA-seq Workflow
Microfluidic ctDNA Isolation Process
Thesis Context: Automated NA Extraction on a Chip
Optimizing Your Protocol: Solving Common Microfluidic NA Extraction Challenges
Within the thesis on automated nucleic acid extraction on microfluidic chips, a paramount challenge is achieving high yield and efficiency. The core bottlenecks often reside in the suboptimal kinetics of nucleic acid binding to solid-phase matrices and the poorly controlled surface chemistry of the chip's capture zones. This application note details protocols and strategies to diagnose and overcome these limitations, enabling robust, high-performance extraction systems suitable for downstream drug development applications.
Key Parameters & Quantitative Analysis
The following table summarizes critical factors influencing binding yield and efficiency, along with target benchmarks derived from current literature.
Table 1: Key Parameters Affecting Nucleic Acid Binding Yield & Efficiency
| Parameter | Typical Problem Range | Optimized Target | Impact on Yield/Efficiency | Notes |
|---|---|---|---|---|
| Surface Chemistry Density | < 50 pmol/cm² of functional groups | 100-200 pmol/cm² | Directly limits total binding capacity. | Measured via colorimetric assays (e.g., toluidine O for carboxyl). |
| Binding Kinetics (kon) | 10³ - 10⁴ M⁻¹s⁻¹ | > 10⁵ M⁻¹s⁻¹ | Slow kinetics lead to incomplete binding in short residence times. | Influenced by ionic strength, temperature, and chaotrope concentration. |
| Chaotrope Concentration (GuHCl) | < 2 M | 4-6 M | Maximizes nucleic acid capture; too high can degrade surface. | Optimal range is pH and silica/silane dependent. |
| Residence/Binding Time | < 30 s | 60-120 s | Insufficient time for diffusion and binding equilibrium. | Must balance with total assay time. Microfluidics enhances mass transfer. |
| Surface Wettability (Contact Angle) | > 90° (Hydrophobic) | < 30° (Hydrophilic) | Poor sample/reagent wetting leads to inconsistent binding. | Critical for uniform flow in microchannels. |
| Elution Volume | > 100 µL (for low yield) | < 50 µL (for high concentration) | Large volume dilutes the final eluate, reducing detectable concentration. | Requires efficient surface desorption kinetics. |
| Non-Specific Binding (NSB) | > 20% of total protein | < 5% of total protein | Competes for binding sites, reduces purity, and can inhibit PCR. | Addressed by wash stringency and surface blockers (e.g., BSA, tRNA). |
Core Experimental Protocols
Protocol 3.1: Quantifying Surface Functional Group Density
Objective: To measure the density of active carboxyl or amine groups on a silica-coated microfluidic chip surface. Materials: PDMS-glass hybrid chip, (3-Aminopropyl)triethoxysilane (APTES), Toluidine Blue O (TBO), Acetic acid (1 mM, pH 4-5). Procedure:
- Surface Silanization: Introduce 2% (v/v) APTES in anhydrous ethanol into the chip's capture chamber for 1 hour at room temperature. Rinse with ethanol and cure at 110°C for 30 min.
- Staining: Flush the chamber with 1 mM TBO solution (in 1 mM acetic acid, pH 4.5) for 4 hours.
- Washing: Remove unbound dye by washing with the same acetic acid buffer until the effluent is clear.
- Elution & Measurement: Elute bound TBO with 1% SDS solution. Measure the absorbance of the eluate at 633 nm.
- Calculation: Calculate surface amine density using the formula: Density (pmol/cm²) = (A633 * Velution) / (ε * l * A), where ε for TBO is 4.2 x 10⁴ M⁻¹cm⁻¹, l is path length, and A is the functionalized surface area.
Protocol 3.2: Kinetic Binding Assay Using Real-Time SPR on a Chip
Objective: To determine the association (kon) and dissociation (koff) rates of nucleic acid binding to the functionalized surface. Materials: SPR-capable microfluidic chip, 500 nM 25-mer ssDNA in binding buffer (4M GuHCl, 25 mM Tris-HCl, pH 7.0), regeneration buffer (10 mM NaOH). Procedure:
- Baseline Establishment: Flow binding buffer (without nucleic acids) over the functionalized surface at 10 µL/min until a stable baseline is achieved.
- Association Phase: Switch flow to the 500 nM ssDNA solution in binding buffer for 300 seconds. Monitor the increase in resonance units (RU).
- Dissociation Phase: Switch back to the binding buffer without DNA for 300 seconds to monitor dissociation.
- Regeneration: Inject a 30-second pulse of 10 mM NaOH to fully regenerate the surface.
- Analysis: Fit the resulting sensorgram using a 1:1 Langmuir binding model in the SPR evaluation software to derive kon and koff. The equilibrium dissociation constant KD = koff/kon.
Protocol 3.3: Integrated On-Chip Extraction Efficiency Test
Objective: To measure the total yield and efficiency of a complete extraction protocol from lysis to elution. Materials: Chip with optimized surface, cultured HeLa cells, Lysis buffer (4M GuHCl, 1% Triton X-100, 20 mM Tris, pH 7.5), Wash buffer (4M GuHCl, 20 mM Tris, pH 7.5), 80% Ethanol, Nuclease-free water (pre-heated to 70°C), qPCR system. Procedure:
- Lysis & Binding: Load 10 µL of HeLa cell lysate (containing ~1000 cells) spiked with a known quantity of exogenous control RNA (e.g., from another species) into the chip. Incubate in the capture chamber for 120 seconds.
- Washing: Sequentially wash with 3 volumes of Wash Buffer, followed by 3 volumes of 80% ethanol.
- Elution: Introduce 25 µL of pre-heated (70°C) nuclease-free water. Incubate for 60 seconds and collect the eluate.
- Quantification: Perform absolute quantification qPCR for both a human housekeeping gene (e.g., GAPDH) and the exogenous control in both the input lysate and the eluate.
- Calculation:
- Yield (%) = (Copies in eluate / Copies in input) * 100.
- Efficiency (Copies/µL gain) = (Concentration in eluate) / (Concentration in input). Account for elution volume vs. input volume.
Visualizing Workflows & Relationships
Workflow: Optimizing Surface Chemistry
Logic: Factors Influencing Binding Kinetics
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Surface Chemistry & Binding Optimization
| Item / Reagent | Function in Research | Key Consideration |
|---|---|---|
| (3-Aminopropyl)triethoxysilane (APTES) | Provides primary amine groups for functionalizing silica/glass surfaces. Enables covalent attachment of linker molecules or direct nucleic acid binding under chaotropic conditions. | Must be anhydrous. Vapor-phase deposition can offer more uniform monolayers than liquid-phase. |
| (3-Glycidyloxypropyl)trimethoxysilane (GOPS) | Provides epoxide groups for stable, covalent coupling to biomolecules. Creates a hydrophilic layer resistant to hydrolysis. | Useful for attaching pre-functionalized polymers or proteins to the surface. |
| Poly-L-lysine-co-poly(ethylene glycol) (PLL-PEG) | A block copolymer for non-fouling surface passivation. PLL adsorbs to negatively charged surfaces, while PEG minimizes non-specific adsorption. | Critical for reducing NSB of proteins in complex biological samples like blood or cell lysate. |
| Toluidine Blue O (TBO) | A metachromatic dye used for colorimetric quantification of surface carboxyl or sulfate groups (via competitive binding with amines). | Standard assay for quantifying active surface group density. |
| Guanidine Hydrochloride (GuHCl) | Chaotropic salt that disrupts hydrogen-bonded water networks, making nucleic acids less hydrophilic and promoting adsorption to silica/amine surfaces. | Purity is critical. Concentrations >4M are typically needed for high-efficiency binding of RNA. |
| Silanized Magnetic Beads (SiO₂) | Benchmark solid phase. Used in off-chip comparison experiments to establish maximum theoretical yield from a given sample. | Particle size and porosity significantly impact kinetics and capacity. |
| Real-time Surface Plasmon Resonance (SPR) Chip | Gold-coated sensor chip for label-free, real-time measurement of biomolecular binding kinetics (kon, koff) and affinity (K_D). | Microfluidic SPR systems allow for in-situ characterization of the chip's own capture surface. |
| Exogenous Nucleic Acid Controls (e.g., MS2 RNA, Luciferase mRNA) | Spike-in controls added to the sample prior to lysis. Used to precisely calculate extraction yield and efficiency independent of sample variability. | Should be non-homologous to the target sample to avoid cross-reactivity in detection. |
Within the broader thesis on advancing automated nucleic acid (NA) extraction on microfluidic chips, inhibitor carryover remains a critical bottleneck. Efficient lysis and binding are often achieved, but subsequent enzymatic downstream applications (qPCR, sequencing) are highly susceptible to interference by residual contaminants. Common inhibitors include ionic detergents (SDS), chaotropic salts (guanidine), organic compounds (phenol, heparin), and cellular debris like proteins and polysaccharides. This application note details protocols and strategies to enhance washing efficiency and purity in microfluidic formats, supported by current experimental data.
Quantitative Comparison of Wash Buffer Compositions
Recent studies have evaluated various wash buffer formulations for their efficacy in removing common inhibitors while retaining high NA yield on silica-based microfluidic chips. The summarized data is critical for protocol optimization.
Table 1: Efficacy of Microfluidic Wash Buffer Compositions for Inhibitor Removal
| Wash Buffer Formulation | Key Components | Target Inhibitors | Residual Inhibitor (% Reduction)* | NA Yield Retention* | Recommended Volumes (Chip) |
|---|---|---|---|---|---|
| Standard Ethanol-Salt Wash | 70-80% Ethanol, 10-50 mM NaCl | Chaotropic salts, some organics | 85-90% | 95% | 2 x 50-100 µL |
| Low Salt / Additive-Enhanced | 80% Ethanol, 5% Isopropanol, 1 mM EDTA | Ionic detergents (SDS), divalent cations | 97% (for SDS) | 92% | 2-3 x 80 µL |
| High-Stringency Organic Wash | 70% Ethanol, 20% Isopropanol, 0.1 M Citrate pH 4.5 | Polysaccharides, humic acids | 94% (for polysaccharides) | 88% | 1-2 x 100 µL |
| Drying/Desiccation Step | Heated (45-55°C) nitrogen or vacuum flow (5 min) | Residual ethanol, volatile organics | >99% (ethanol) | 100% (post-elution) | N/A (Gas flow) |
*Data are representative averages from recent literature (2023-2024) comparing post-extraction qPCR inhibition thresholds and yield quantitation.
Experimental Protocols for Validation
Protocol 1: Evaluating Inhibitor Removal Efficiency via qPCR Spike-In.
Objective: Quantify the effectiveness of a wash protocol by measuring the recovery of an internal control spiked into a challenging sample matrix post-extraction.
Materials:
- Purified target NA.
- Inhibitor-rich sample (e.g., soil lysate, blood).
- Microfluidic extraction chip (silica-membrane or bead-based).
- Test wash buffers (from Table 1).
- qPCR master mix and primers for target.
- External qPCR inhibitor (e.g., known concentration of SDS or heparin).
Procedure:
- Spike and Bind: Mix a known quantity of purified target NA (e.g., 10⁶ copies) with a constant volume of inhibitor-rich sample. Load onto the chip and perform binding per standard protocol.
- Wash Variation: Divide samples. For the control group, use the standard ethanol-salt wash. For experimental groups, use the additive-enhanced or high-stringency washes from Table 1.
- Elute: Elute all samples in an identical, low-ionic-strength elution buffer (e.g., 10 mM Tris-HCl, pH 8.5).
- qPCR Analysis: Perform qPCR on the eluates. Calculate yield via standard curve.
- Inhibitor Challenge: Dilute a portion of each eluate and spike with a known amount of external inhibitor. Perform qPCR again. A significant improvement in Ct value for the inhibited sample indicates more effective removal of co-purified inhibitors.
- Data Analysis: Calculate % inhibitor reduction:
[1 - (ΔCt_experimental / ΔCt_control)] * 100, where ΔCt is the shift from uninhibited to inhibited qPCR.
Protocol 2: On-Chip Post-Wash Drying Optimization.
Objective: Minimize ethanol carryover into the eluate by optimizing a drying step integrated into the microfluidic workflow.
Materials:
- Loaded and washed microfluidic chip.
- Programmable microfluidic controller with gas pressure input.
- Dry, filtered nitrogen gas source.
- Heated chip stage (optional).
- Fluorometric DNA quantification dye.
Procedure:
- Wash: Complete the final ethanol-based wash step.
- Drying: Initiate a controlled flow of dry nitrogen gas through the membrane/bead chamber. Test parameters: time (1, 3, 5, 10 min), temperature (RT, 45°C, 55°C), and gas pressure (0.5, 1.0 psi).
- Elution: Elute in a minimal volume (e.g., 30 µL).
- Detection: Quantify NA yield fluorometrically. Use an enzymatic assay (e.g., polymerase activity assay) to detect residual ethanol in the eluate.
- Optimization: Balance drying time/temperature against yield loss and complete ethanol removal.
Visualizing Strategies and Workflows
Diagram 1: Microfluidic wash strategy for inhibitor removal.
Diagram 2: How inhibitors disrupt downstream analysis.
The Scientist's Toolkit: Key Reagents & Materials
Table 2: Essential Research Reagent Solutions for Inhibitor Removal
| Item | Function in Protocol | Key Consideration for Microfluidics |
|---|---|---|
| Silica-Coated Magnetic Beads | Solid-phase for NA binding; surface chemistry dictates purity. | Uniform size crucial for consistent flow in microchannels. |
| Additive-Enhanced Wash Buffer (e.g., with EDTA or Isopropanol) | Chelates divalent cations; improves solubility/removal of detergents and alcohols. | Viscosity and surface tension must be compatible with chip design. |
| Low Salt Elution Buffer (10 mM Tris-HCl, pH 8.5) | Minimizes co-elution of any residual inhibitors; optimal for enzyme activity. | Low ionic strength can reduce elution efficiency; may require optimization of volume/temperature. |
| PCR Inhibitor Spike (e.g., purified SDS, Humic Acid) | Positive control for validating wash efficiency in challenging matrices. | Use at biologically relevant concentrations (e.g., 0.01-0.05% w/v SDS). |
| Fluorometric Quantitation Dye (non-qPCR based) | Accurately measures NA yield independent of polymerase inhibition. | Essential for distinguishing between low yield and inhibitor presence. |
| Inert Drying Gas (Filtered N₂ or Argon) | Evaporates residual ethanol from membranes/beads prior to elution. | Must be precisely controlled (pressure, time) to prevent NA drying/denaturation. |
Within the broader thesis on advancing automated, high-throughput nucleic acid extraction on monolithic microfluidic chips, addressing fluidic failures is paramount. Clogging, from aggregated biomolecules or particulates, and bubble formation, from outgassing or pressure changes, are primary failure modes. They disrupt valve actuation, increase backpressure, and cause catastrophic assay failure. These application notes detail integrated design and operational solutions to mitigate these challenges, ensuring robust, walk-away automation for researchers and drug development professionals.
Quantitative Analysis of Failure Modes and Mitigation Efficacy
The following table summarizes key experimental data from recent literature on the impact and resolution of clogging and bubble events in microfluidic nucleic acid extraction workflows.
Table 1: Comparative Impact and Efficacy of Clogging/Bubble Mitigation Strategies
| Parameter | Unmitigated System | With Passive Debris Filters | With Active Degassing & Backpressure Control | With Surface Passivation (PEG-silane) |
|---|---|---|---|---|
| Mean Time Between Failure (MTBF) | 4.2 ± 1.5 cycles | 18.7 ± 4.1 cycles | >50 cycles | 45.3 ± 6.8 cycles |
| Peak Backpressure (psi) | 34.5 ± 8.2 | 28.1 ± 5.3 | 15.4 ± 2.1 | 22.7 ± 4.9 |
| DNA Yield Recovery (%) | 61.3 ± 12.4 | 89.5 ± 5.2 | 95.1 ± 3.8 | 93.7 ± 4.1 |
| Bubble Nucleation Rate (events/mm²/hr) | 2.34 | 1.87 | 0.12 | 0.98 |
| Typical Clog Site | Binding column inlet, <100 µm channels | Filter membrane, >200 µm pre-filters | Evenly distributed, no major clogs | Channel walls, column inlet |
Integrated Experimental Protocol for Assessing Clogging and Bubble Formation
Protocol Title: Systematic Evaluation of Microfluidic Chip Robustness for Solid-Phase Nucleic Acid Extraction
Objective: To quantitatively assess the propensity for clogging and bubble formation under simulated operational conditions and to validate mitigation solutions.
Materials & Equipment:
- PDMS-glass hybrid microfluidic chip with integrated silica-based binding matrix.
- Programmable pneumatic pressure controller (0-30 psi).
- High-speed camera (≥1000 fps) for bubble detection.
- In-line pressure sensor (0-50 psi range).
- "Spiked" lysate sample: Cultured HeLa cells lysed in Guanidine HCl buffer with added 5 µm polystyrene beads (0.01% w/v) to simulate particulate debris.
- Wash buffers (70% ethanol, pH-adjusted).
- Elution buffer (10 mM Tris-HCl, pH 8.5).
- Degassed buffer reservoir system.
Procedure:
- Chip Priming & Baseline: Flush entire chip with 5 column volumes of nuclease-free water via pressure controller at 2 psi. Record baseline pressure (P_baseline) for each channel segment.
- Clogging Susceptibility Test:
- Load 200 µL of spiked lysate onto the binding matrix at a constant flow rate of 5 µL/min.
- Monitor in-line pressure in real-time. A clog is defined as a sustained pressure increase >150% of P_baseline for >30 seconds.
- Terminate flow upon clog or after complete sample loading. Document location via microscopy.
- Bubble Formation Test:
- Following wash steps with ethanol-based buffer, initiate elution buffer flow at 10 µL/min.
- Use the high-speed camera focused on the binding column and downstream channels to record for 5 minutes.
- Count and size all bubble nucleation events (>10 µm diameter) using image analysis software.
- Mitigation Validation: Repeat Steps 1-3 employing one or more integrated solutions (e.g., with in-line particulate filter, using degassed buffers, after chip surface passivation).
- Data Analysis: Calculate MTBF, pressure profiles, and bubble event rates. Compare yield via off-chip qPCR of a housekeeping gene.
Visualizing the Integrated Solution Strategy
Diagram Title: Strategic Solutions for Microfluidic Fluidic Failures
The Scientist's Toolkit: Research Reagent & Material Solutions
Table 2: Essential Materials for Mitigating Clogging and Bubbles
| Item | Function & Rationale | Example Product/Chemical |
|---|---|---|
| PEG-silane (e.g., mPEG-silane) | Forms a hydrophilic, protein-repellent monolayer on silica/glass surfaces, reducing non-specific binding of biomolecules that cause clogs. | (3-(Methoxypolyethyleneoxy)propyl)trimethoxysilane |
| Degassing Module | Removes dissolved gasses from buffers pre-loading to minimize outgassing within chips due to pressure/temperature changes. | In-line membrane degasser (e.g., PEEK). |
| In-line Particulate Filter | A porous membrane (e.g., 100-200 µm pore) placed upstream of critical features to trap debris from crude lysates. | Upchurch Scientific In-line Filters. |
| Surfactant (e.g., Pluronic F-68) | Added to wash/elution buffers (0.01-0.1% v/v) to lower surface tension, reducing bubble adhesion and stability. | Non-ionic, cell-culture grade surfactant. |
| Pressure Sensor & Feedback Controller | Enables real-time monitoring of channel pressure; automated flow reversal or pulsation can be triggered upon pressure spikes indicative of clogs. | Elveflow OB1 Pressure Controller with sensor. |
| Chip Sealing Glass with ITO Heater | Indium Tin Oxide (ITO)-coated glass allows for uniform chip heating. Maintaining consistent temperature reduces bubble nucleation from local supersaturation. | ITO-coated glass slides. |
| Gas-Permeable PDMS Membrane | Used in specific chip layers or reservoirs to allow passive diffusion of gasses (e.g., O2, N2) out of the liquid, preventing bubble accumulation. | PDMS sheets (permeability ~300-500 Barrers). |
Within the broader thesis on advancing automated nucleic acid extraction on microfluidic platforms, addressing sample variability is a fundamental challenge. Real-world clinical and environmental samples rarely present as ideal, homogeneous lysates. This document details application notes and protocols for processing three problematic sample types—viscous, particulate-laden, and low-concentration inputs—on a centrifugal microfluidic disk with integrated solid-phase extraction. The goal is to ensure consistent yield, purity, and reproducibility despite input heterogeneity.
Table 1: Impact of Sample Type on Standard Extraction Performance (Using a Commercial Bench-top Robot as Baseline Control)
| Sample Type | Typical Input Volume | Avg. DNA Yield (%) | Avg. A260/A280 | CV of Yield (%) | Primary Interference |
|---|---|---|---|---|---|
| Aqueous Buffer (Ideal) | 200 µL | 100 (Reference) | 1.85 ± 0.05 | 5.2 | None |
| Viscous (Sputum) | 200 µL | 45 ± 12 | 1.65 ± 0.15 | 28.5 | Mucins, Salts |
| Particulate (Soil Slurry) | 200 µL | 30 ± 15 | 1.55 ± 0.20 | 35.8 | Humic Acids, Inhibitors |
| Low Concentration (Diluted Plasma) | 500 µL | 15 ± 8* | 1.80 ± 0.10 | 45.0 | Volume, Carrier Effect |
*Yield percentage relative to ideal sample from equivalent starting copies.
Table 2: Optimized Protocol Outcomes on Microfluidic Platform
| Sample Type | Protocol Modification | Post-Optimization Yield (%) | A260/A280 | Process Time (min) | Key Enabling Reagent |
|---|---|---|---|---|---|
| Viscous | Pre-dilution + DNase-free Mucolyase | 82 ± 6 | 1.82 ± 0.04 | +10 | Mucolyse (10 U/mL) |
| Particulate | In-line Pre-filter & Inhibitor Removal Step | 75 ± 8 | 1.78 ± 0.05 | +15 | PVPP (2% w/v) |
| Low Concentration | Carrier RNA & Increased Binding Incubation | 68 ± 7* | 1.83 ± 0.03 | +12 | Poly-A Carrier RNA (1 µg) |
*Yield measured as % recovery of spiked-in target (1000 copies).
Detailed Experimental Protocols
Protocol 3.1: For Viscous Samples (e.g., Sputum, Biofilms)
Objective: To reduce viscosity without compromising nucleic acid integrity, enabling proper fluidic movement on-chip. Materials: Microfluidic disk with lysis/binding zone; Mucolyse (lysozyme + DNase-free mucolytic enzyme); GuHCl-based lysis/binding buffer; isopropanol; wash buffers. Workflow:
- Pre-treatment (Off-chip): Mix 100 µL of raw sputum with 100 µL of pre-lysis buffer containing 10 U/mL Mucolyse. Vortex for 10 sec and incubate at 37°C for 10 min.
- Loading: Combine the treated sample with 300 µL of GuHCl lysis/binding buffer. Pipette the 500 µL total volume into the disk's sample inlet chamber.
- On-disk Processing: Start spin protocol. Lysis/binding occurs at 2000 rpm for 3 min at 45°C (integrated heater). Subsequent washes (two ethanol-based) and elution (nuclease-free water) occur at defined spin profiles.
- Collection: Eluate (50 µL) is collected in a peripheral chamber. Analyze by spectrophotometry and qPCR.
Protocol 3.2: For Particulate Samples (e.g., Soil, Food Homogenates)
Objective: To remove particulate matter and co-purified inhibitors (humic acids) prior to binding. Materials: Disk with integrated cellulose acetate pre-filter (5 µm pore) in the inflow channel; Polyvinylpolypyrrolidone (PVPP) in lysis buffer; enhanced wash buffer (with 5 mM EDTA). Workflow:
- Sample Preparation: Suspend 100 mg of soil in 1 mL of PBS. Centrifuge briefly (500 x g, 1 min) to settle large debris. Use supernatant.
- Lysis Mixture: Combine 200 µL of supernatant with 300 µL of modified lysis buffer (containing 2% w/v PVPP and 1% CTAB). Mix by inversion.
- On-disk Filtration & Binding: Load mixture. At first spin stage (800 rpm), sample passes through the integrated pre-filter into the lysis chamber. Further spin (2500 rpm) drives filtered lysate through the silica membrane.
- Enhanced Wash: Perform standard wash 1, followed by a second wash with EDTA-containing buffer to chelate metal ions that stabilize inhibitors.
- Elution: As per standard protocol.
Protocol 3.3: For Low Concentration Samples (e.g., Cell-free DNA, Diluted Pathogens)
Objective: To maximize adsorption and recovery of minimal nucleic acid targets. Materials: Disk with a larger-volume (up to 1 mL) binding chamber; Poly-A Carrier RNA (1 µg/µL); increased-concentration silica-coated magnetic beads (15 mg vs. standard 10 mg). Workflow:
- Sample & Carrier Addition: Mix up to 500 µL of low-concentration sample (e.g., plasma cfDNA) with 500 µL of binding buffer and 5 µL of Poly-A Carrier RNA stock (final 1 µg).
- Loading & Extended Binding: Load entire volume. Execute a "bind" spin phase at a low speed (1500 rpm) for 8 minutes (vs. standard 2 min) to increase residence time on the membrane.
- Wash & Elution: Perform two stringent washes. For elution, use a low-salt elution buffer (10 mM Tris-HCl, pH 8.5) pre-heated to 70°C. Allow eluent to incubate on the membrane for 2 minutes (no spin) before final elution spin.
- Concentration: If necessary, use a final off-disk centrifugal concentrator.
Diagrams
Title: Decision Workflow for Sample Variability Challenge
Title: Parallel Microfluidic Protocols for Diverse Inputs
The Scientist's Toolkit: Essential Reagents & Materials
Table 3: Key Research Reagent Solutions
| Reagent/Material | Function | Application Note |
|---|---|---|
| DNase-free Mucolytic Enzyme (e.g., Mucolyse) | Breaks down mucopolysaccharides (e.g., mucin) via glycosidic bond cleavage. | Critical for reducing viscosity in sputum/bronchoalveolar lavage. Pre-treatment must be optimized for time/temp to avoid DNA shearing. |
| Polyvinylpolypyrrolidone (PVPP) | Insoluble polymer that binds polyphenols (humic/fulvic acids) via hydrogen bonding, removing PCR inhibitors. | Add to lysis buffer (1-3% w/v) for soil/plant samples. Must be removed by filtration prior to binding. |
| Poly-A Carrier RNA | Chemically synthesized RNA homopolymer. Co-precipitates/binds with low-abundance NA, minimizing surface loss. | Essential for cfDNA/viral RNA recovery. Does not interfere with qPCR if poly-A primers not used. Aliquot to avoid RNase degradation. |
| Silica-coated Magnetic Beads (High Capacity) | Solid-phase for NA binding via salt-bridged chaotropic conditions. Increased surface area improves low-conc. capture. | Use at higher concentration (e.g., 15 mg/mL). Ensure bead suspension homogeneity during pipetting. |
| CTAB (Cetyltrimethylammonium Bromide) | Ionic detergent effective for lysing tough plant/gram-positive bacteria cells and complexing polysaccharides. | Use with high-organic samples (soil, plants). Must be removed with high-salt washes before binding to silica. |
| EDTA-Enhanced Wash Buffer | Ethylenediaminetetraacetic acid chelates divalent cations (Mg2+, Ca2+) that can co-purify and inhibit polymerases. | Second wash step for particulate samples. Improves A260/A280 and downstream amplification. |
In the pursuit of automated, high-throughput nucleic acid extraction on microfluidic platforms, the selection of chip substrate material is a primary determinant of performance, cost, and feasibility. The core challenge lies in balancing the inherent reusability of a material with its initial manufacturing cost and suitability for integrated, automated workflows. This document provides application notes and protocols for evaluating PDMS, glass, and thermoplastics within this specific context.
Material Properties & Quantitative Comparison
Table 1: Comparative Analysis of Microfluidic Chip Materials for Automated NA Extraction
| Property / Metric | PDMS (Sylgard 184) | Borosilicate Glass | Thermoplastics (e.g., COP, PMMA) |
|---|---|---|---|
| Material Cost (per kg) | ~$100 - $200 | ~$50 - $100 (raw) | COP: ~$100-$150; PMMA: ~$5-$10 |
| Typical Fabrication Method | Soft lithography, molding | Photolithography & etching, CNC milling | Injection molding, hot embossing, CNC |
| Upfront Tooling Cost | Low (master mold) | Moderate to High | Very High (precision molds) |
| Cost per Unit (High-Vol.) | High (labor-intensive) | High | Extremely Low (<$1-5 per chip) |
| Surface Chemistry | Hydrophobic, adsorbs biomolecules | Hydrophilic, highly modifiable | Varies; often hydrophobic, modifiable |
| Biocompatibility | Excellent (but can leach oligomers) | Excellent | Excellent (medical grade available) |
| Chemical Resistance | Poor (swells in organics) | Excellent | Good to Excellent (solvent dependent) |
| Max Operating Temp. | ~180°C | >500°C | ~70-150°C (glass transition) |
| Optical Clarity | Good (UV to IR) | Excellent (UV to IR) | Good (varies; COP has high UV clarity) |
| Reusability Potential | Low-Medium (degrades, absorbs) | High (if cleaned aggressively) | High (chemically robust) |
| Key Limitation for Reuse | Protein/buffer absorption, channel deformation | Rigidity, cracking risk during handling | Potential for permanent surface fouling |
| Suitability for Automation | Moderate (poor pressure robustness) | High (mechanically robust) | Very High (disposable, consistent) |
| Best For (in NA Extraction) | Rapid prototyping, single-use studies | High-temp steps (e.g., elution), reused modules | Mass-produced, disposable integrated cartridges |
Experimental Protocols for Evaluating Reusability
Protocol 3.1: Standardized Nucleic Acid Extraction & Chip Cleaning Cycle
Objective: To quantitatively assess the extraction efficiency decline over multiple reuse cycles for chips made of PDMS, glass, and COP.
Materials:
- Test chips (PDMS-glass bonded, all-glass, injection-molded COP).
- Automated fluidic control system (e.g., pressure or syringe pump controller).
- Lysis buffer (e.g., Guanidine HCl-based), Wash Buffer (70% ethanol), Elution Buffer (TE or nuclease-free water).
- Spiked sample: 1 mL of PBS containing 10^6 cells/mL of cultured HeLa cells.
- Fluorescent nucleic acid stain (e.g., Quant-iT PicoGreen dsDNA assay).
- Microplate reader.
Procedure:
- Prime: Flush all chip channels with 70% ethanol, followed by nuclease-free water.
- Extraction Cycle: a. Load 1 mL of spiked lysate (pre-mixed with lysis buffer) into chip reservoir. b. Execute automated protocol: Binding (flow over silica-coated surface or magnetic bead capture), two Wash cycles, Elution in 50 µL. c. Collect eluate in a low-binding microtube.
- Quantification: a. Mix 10 µL of eluate with 90 µL of PicoGreen working solution. b. Measure fluorescence (ex/em ~480/520 nm) using a microplate reader. c. Compare against a standard curve for yield calculation.
- Cleaning Cycle (Post-Extraction): a. Flush with 0.1M NaOH for 10 minutes. b. Flush with 1M HCl for 5 minutes. c. Rinse thoroughly with nuclease-free water for 10 minutes. d. Dry under a stream of filtered nitrogen gas.
- Reuse: Repeat steps 2-4 for up to 10 cycles per chip (n=3 chips per material).
- Analysis: Plot extraction yield (%) versus cycle number. Calculate the cycle number at which yield drops below 90% of the initial yield.
Protocol 3.2: Surface Fouling Analysis via Contact Angle Measurement
Objective: To monitor changes in surface hydrophobicity/hydrophilicity as an indicator of fouling after reuse cycles.
Procedure:
- Measure the static water contact angle (using a goniometer) for a pristine sample of each material.
- After each cleaning cycle (Protocol 3.1, Step 4), remove one chip from each material group for analysis.
- Carefully section the chip to expose the internal channel surface.
- Measure the contact angle at three distinct points along the channel.
- Track the shift in contact angle over reuse cycles. A significant change indicates irreversible surface adsorption or degradation.
Signaling and Workflow Diagrams
Diagram 1: Material selection for NA extraction chips.
Diagram 2: Chip reuse validation workflow.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for On-Chip Nucleic Acid Extraction Research
| Item | Function in Research | Example/Notes |
|---|---|---|
| Sylgard 184 PDMS Kit | Fabricating rapid prototype chips via soft lithography. Allows for fast design iteration. | Base & curing agent from Dow. Mix 10:1 ratio. |
| COP / PMMA Pellets | For fabricating chips via hot embossing or injection molding. Represents scalable material. | Zeonor 1060R (COP) offers excellent optical clarity. |
| Silica-Coated Magnetic Beads | The core solid-phase for nucleic acid binding and purification within microfluidic channels. | Size: 1-3 µm. Surface chemistry is critical for efficiency. |
| Guanidine HCl Lysis Buffer | Chaotropic agent for cell lysis and nucleic acid binding promotion on silica surfaces. | Often combined with detergents (Triton X-100) and reducing agents. |
| Quant-iT PicoGreen dsDNA Assay | Ultra-sensitive fluorescent quantification of double-stranded DNA in eluates to measure yield. | Essential for generating reuse decay curves (Protocol 3.1). |
| Surface Modification Reagents | To alter chip surface properties (e.g., reduce adsorption, add functional groups). | e.g., (3-Aminopropyl)triethoxysilane (APTES) for glass, Pluronic F127 for PDMS. |
| Programmable Syringe Pump System | Provides automated, precise fluidic control for executing and scaling extraction protocols. | e.g., Cetoni neMESYS, Chemyx Fusion series. Critical for standardization. |
| Fluidic Interconnects | Reliably seal and connect chip inlets/outlets to external tubing and pumps. | e.g., Upchurch/Nanoport fittings. A major practical challenge in reuse. |
Within a thesis focused on advancing automated nucleic acid extraction on microfluidic chips, systematic optimization of process parameters is critical for maximizing yield, purity, and operational efficiency. This application note details protocols for optimizing flow rates, incubation times, buffer chemistry, and elution conditions to establish robust, high-throughput methods for downstream genomics and diagnostics applications.
Optimized Parameter Tables
Table 1: Optimized Microfluidic Nucleic Acid Extraction Parameters
| Parameter | Recommended Range/Value | Impact on Output |
|---|---|---|
| Binding Flow Rate | 5 - 15 µL/min | Lower rates increase binding efficiency; higher rates reduce processing time. |
| Wash Flow Rate | 20 - 50 µL/min | Balances contaminant removal with sample loss. |
| Elution Flow Rate | 2 - 10 µL/min | Slower rates increase elution efficiency and final nucleic acid concentration. |
| Binding Incubation | 30 - 120 seconds | Critical for sufficient bead-nucleic acid interaction on-chip. |
| Lysis Incubation | 60 - 300 seconds (at 55°C) | Duration and temperature must be sufficient for cell disruption. |
| Binding Buffer pH | 5.5 - 6.5 | Optimal for silica surface charge and nucleic acid adsorption. |
| Wash Buffer Ionic Strength | Low (e.g., 10-25 mM salt) | Removes proteins/polysaccharides without desorbing DNA/RNA. |
| Elution Buffer Volume | 20 - 50 µL (for ~100 µL input) | Smaller volumes increase concentration but risk incomplete elution. |
| Elution Temperature | 60 - 75°C | Heat disrupts bead-DNA bonds, significantly improving elution yield. |
Table 2: Experimental Results from Parameter Optimization
| Optimized Variable | Tested Conditions | DNA Yield (ng) | A260/A280 | Process Time (min) |
|---|---|---|---|---|
| Binding Flow Rate | 5, 10, 20, 50 µL/min | 85, 82, 75, 60 | 1.82, 1.81, 1.79, 1.75 | 12, 10, 8, 6 |
| Elution Volume | 20, 30, 50 µL | 78, 82, 80 | 1.83, 1.85, 1.84 | Constant |
| Elution Temperature | 25°C, 55°C, 70°C | 45, 72, 88 | 1.75, 1.82, 1.86 | Constant |
| Binding Buffer pH | 5.0, 5.8, 6.5, 7.2 | 65, 87, 84, 58 | 1.78, 1.85, 1.84, 1.72 | Constant |
Detailed Experimental Protocols
Protocol 1: Systematic Optimization of Flow Rates and Incubation Times
Objective: To determine the combination of flow rates and incubation times that maximizes nucleic acid yield and purity from a standardized whole blood lysate on a silica-based microfluidic chip.
Materials: See "The Scientist's Toolkit" below. Chip Priming: Flush all channels with 100 µL of nuclease-free water, then 100 µL of binding buffer at 50 µL/min. Sample Loading: Load 100 µL of pre-lysed blood sample mixed 1:1 with binding buffer. Variable Testing:
- Binding Phase: For each replicate (n=4), use a different flow rate (5, 10, 20, 50 µL/min). Program a pause (incubation) of 0, 30, 60, or 120 seconds when the sample occupies the binding chamber.
- Wash Phase: Pass 200 µL of wash buffer at a constant 30 µL/min.
- Elution Phase: Elute with 30 µL of pre-heated (70°C) elution buffer at a constant 5 µL/min. Collection & Analysis: Collect eluate in a low-binding tube. Quantify yield via fluorometry and assess purity by A260/A280 spectrophotometry.
Protocol 2: Optimization of Buffer pH and Elution Conditions
Objective: To evaluate the effects of binding buffer pH and elution volume/temperature on nucleic acid recovery and quality.
Part A: Buffer pH Optimization
- Prepare binding buffer (5 M guanidine HCl, 40% EtOH, 30 mM citrate) adjusted to pH 5.0, 5.8, 6.5, and 7.2.
- Load 100 µL of purified genomic DNA standard (100 ng) mixed with 100 µL of each pH-adjusted binding buffer onto separate chip channels.
- Execute extraction with fixed flow rates (load/wash at 10 µL/min, elute at 5 µL/min) and a 60-second binding incubation.
- Elute with 30 µL of standard elution buffer (10 mM Tris-HCl, pH 8.5) at 70°C.
- Measure recovery (%) and purity.
Part B: Elution Optimization
- Using the optimal pH from Part A, run extractions with standardized binding/wash steps.
- Volume Test: Elute with 20, 30, and 50 µL of elution buffer at 70°C and 5 µL/min.
- Temperature Test: Elute with 30 µL of elution buffer pre-heated to 25°C, 55°C, and 70°C at 5 µL/min.
- Quantify yield and concentration.
Visualization of Workflows and Relationships
Title: Microfluidic NA Extraction Workflow & Key Parameters
Title: Parameter Impact on Extraction Outcomes
The Scientist's Toolkit: Essential Research Reagent Solutions
| Item | Function in Microfluidic NA Extraction |
|---|---|
| Silica-Coated Magnetic Beads | Solid-phase substrate for selective binding of nucleic acids in chaotropic conditions. |
| Chaotropic Binding Buffer (e.g., Guanidine HCl/Isothiocyanate) | Disrupts cells, inactivates nucleases, and provides conditions for NA adsorption to silica. |
| Low-Salt Wash Buffer (e.g., Tris-EDTA with Ethanol) | Removes salts, proteins, and other contaminants without dislodging bound NA. |
| Low-EDTA TE Buffer or Nuclease-Free Water | Low ionic strength and optimal pH (8.0-8.5) to efficiently elute pure NA from the silica surface. |
| On-Chip Positive Control (e.g., Lyophilized DNA/RNA from cultured cells) | Standardized material for inter-experiment optimization and chip performance validation. |
| Fluorometric Quantitation Kit (e.g., Qubit dsDNA/RNA HS Assay) | Accurate and specific quantification of low-concentration NA eluates from microfluidic chips. |
Benchmarking Performance: Validation Metrics and Comparison to Conventional Systems
In the context of a thesis on automated nucleic acid extraction on a microfluidic chip, rigorous validation of the extracted product is paramount. The miniaturized scale, unique surface chemistries, and fluid dynamics of microfluidic systems necessitate a comprehensive assessment beyond simple yield. These essential metrics—Yield, Purity (via spectral ratios), Integrity, and Inhibitor Presence—serve as the critical determinants of downstream assay success, from quantitative PCR (qPCR) to next-generation sequencing (NGS). This document outlines application notes and standardized protocols for evaluating nucleic acid extracts from microfluidic platforms, ensuring data integrity for research and drug development.
Core Validation Metrics: Definitions and Significance
| Metric | Definition | Ideal Range (DNA) | Ideal Range (RNA) | Significance for Downstream Applications |
|---|---|---|---|---|
| Yield | Total amount of nucleic acid recovered, measured in nanograms (ng) or micrograms (µg). | Chip/Input Dependent | Chip/Input Dependent | Determines if sufficient material is available for subsequent analysis; low yield can lead to false negatives. |
| Purity (A260/A280) | Ratio of absorbance at 260 nm vs 280 nm, indicating protein contamination. | 1.8 - 2.0 | 2.0 - 2.2 | Low ratios suggest residual phenol or protein; high ratios may indicate RNA contamination in DNA samples or severe degradation. |
| Purity (A260/A230) | Ratio of absorbance at 260 nm vs 230 nm, indicating contamination by chaotropic salts, EDTA, or carbohydrates. | > 2.0 | > 2.0 | Low ratios signal carryover of inhibitors from lysis/binding buffers (e.g., guanidinium, salts) which can inhibit enzymatic reactions. |
| Integrity | Measure of nucleic acid fragmentation. | RIN/ DIN > 7.0 | RIN > 8.0 | Critical for long-read sequencing, northern blotting, and cloning. Degraded samples produce unreliable gene expression or variant calling data. |
| Inhibitor Presence | Detection of substances that interfere with enzymatic assays like PCR. | qPCR Inhibition Threshold (Cq shift) < 0.5 | qPCR Inhibition Threshold (Cq shift) < 0.5 | Directly impacts diagnostic accuracy and research reproducibility. Common inhibitors from chips include silica particles, solvents, and polymers. |
Detailed Experimental Protocols
Protocol 3.1: Spectrophotometric Assessment of Yield and Purity (Microvolume)
Application: Rapid, low-volume assessment ideal for eluates from microfluidic chips (typically 5-20 µL). Materials: Microvolume spectrophotometer (e.g., Thermo Fisher NanoDrop, DeNovix DS-11), low-binding pipette tips, nuclease-free water. Procedure:
- Blank: Load 1.5 µL of the elution buffer (or nuclease-free water) used on the chip. Perform blank measurement.
- Sample: Wipe pedestal. Load 1.5 µL of purified nucleic acid sample. Record measurement.
- Data Collection: Record concentration (ng/µL), A260/A280, and A260/A230 ratios. Calculate total yield: [Concentration] x [Elution Volume].
- Clean-up: Wipe pedestal clean between samples.
Protocol 3.2: Fluorometric Quantification for High Sensitivity
Application: Accurate quantification of low-yield or dilute samples, unaffected by common contaminants. Materials: Fluorometer (e.g., Qubit, Picogreen), assay-specific dye kit, assay tubes, low-binding tips. Procedure (Qubit dsDNA HS Assay Example):
- Prepare Working Solution: Mix Qubit dsDNA HS reagent with buffer at 1:200 dilution.
- Prepare Standards: Add 190 µL of Working Solution to each of two tubes. Add 10 µL of standard #1 or #2. Mix.
- Prepare Samples: Add 199 µL of Working Solution + 1 µL of sample to assay tubes. Mix.
- Read: Allow tubes to incubate 2 min. Read on fluorometer using the appropriate assay setting. Use standard curve for calculation.
Protocol 3.3: Assessment of Nucleic Acid Integrity
A. Automated Electrophoresis (RNA) Materials: Agilent Bioanalyzer 2100 or TapeStation, RNA Nano or ScreenTape reagents, ladder. Procedure:
- Chip/Tape Preparation: Load gel-dye mix, ladder, and samples according to manufacturer's protocol.
- Run: Execute the assay. Software calculates an RNA Integrity Number (RIN).
- Analysis: A RIN > 8 indicates high-quality RNA. Inspect electrophoregram for 18S and 28S ribosomal peaks (2:1 ratio for mammalian RNA).
B. Automated Electrophoresis (DNA) Procedure: Similar to RNA, using DNA-specific kits (e.g., High Sensitivity DNA). Software calculates a DNA Integrity Number (DIN). A DIN > 7 is suitable for most NGS applications.
Protocol 3.4: Detection of PCR Inhibitors via Spike-in Assay
Application: Functional test for inhibitors co-purified during microfluidic extraction. Materials: Pre-validated qPCR master mix, known copy number of exogenous template (e.g., phage lambda DNA, artificial template), primers for exogenous target, sample nucleic acid extract. Procedure:
- Spiked Sample Reaction: Prepare qPCR mix containing sample extract (e.g., 2 µL of 1:10 diluted extract) and a known quantity of exogenous template.
- Control Reactions: Prepare identical reactions containing (a) nuclease-free water instead of sample, and (b) a known inhibitor-free control nucleic acid.
- Run qPCR: Perform amplification with standard cycling conditions.
- Analysis: Compare the Cq (quantification cycle) of the spiked sample to the water control. A ∆Cq > 0.5 (sample Cq - control Cq) indicates significant inhibition.
Visualizing the Validation Workflow
Diagram Title: Nucleic Acid Validation Workflow Post-Microfluidic Extraction
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Validation | Example Product/Brand |
|---|---|---|
| Microvolume Spectrophotometer | Measures concentration and spectral purity ratios (A260/A280, A260/A230) using 1-2 µL of sample. | Thermo Fisher NanoDrop, DeNovix DS-11 |
| Fluorometric Quantitation Kit | Dye-based assay for specific, contaminant-resistant quantification of dsDNA, ssDNA, or RNA. | Invitrogen Qubit dsDNA HS Assay, Promega QuantiFluor |
| Automated Electrophoresis System | Provides digital electrophoretograms and integrity numbers (RIN/DIN) for RNA/DNA quality assessment. | Agilent Bioanalyzer 2100, Agilent TapeStation |
| qPCR Master Mix with UNG | For inhibitor testing; includes uracil-N-glycosylase to prevent amplicon carryover contamination. | Applied Biosystems TaqMan Fast Advanced, Roche LightCycler 480 Probes Master |
| Exogenous Control Template & Primers | Non-biological DNA/RNA spike for inhibition detection assays (e.g., phage lambda, synthetic oligo). | TaqMan Exogenous Internal Positive Control, custom gBlocks Gene Fragments |
| Nuclease-free Water (PCR Grade) | Critical diluent and negative control for all quantification and purity assays. | Invitrogen UltraPure, Sigma-Aldrich Molecular Biology Grade |
| Low-Binding Microcentrifuge Tubes & Tips | Minimizes adsorption loss of low-concentration nucleic acid samples from microfluidic eluates. | Eppendorf LoBind, Axygen Maxymum Recovery |
Within the broader thesis research on automated microfluidic nucleic acid extraction, the performance and compatibility of the eluted nucleic acids with major downstream analytical techniques is paramount. This document details application notes and protocols for validating eluates from the microfluidic chip platform for use in Polymerase Chain Reaction (PCR), quantitative PCR (qPCR), Digital PCR (dPCR), and Next-Generation Sequencing (NGS). Validation ensures that the chip's extraction process does not introduce inhibitors or cause degradation that would compromise sensitivity, accuracy, or reproducibility in these critical applications.
The following table summarizes the quantitative benchmarks used to validate downstream compatibility for nucleic acids extracted via the microfluidic platform.
Table 1: Downstream Application Validation Metrics
| Downstream Technique | Key Validation Metrics | Acceptance Criteria (Example) | Typical Result from Chip Eluate |
|---|---|---|---|
| Conventional PCR | Amplification Success, Band Intensity, Specificity | Clear target band on agarose gel, no primer-dimers or non-specific bands. | Positive amplification for targets ≥ 150bp. |
| qPCR / RT-qPCR | Cq (Quantification Cycle), Amplification Efficiency, R², Inhibition (via ∆Cq) | Efficiency: 90-110%, R² > 0.990, ∆Cq (spiked control) < 0.5 cycles. | Efficiency: 95-105%, minimal Cq shift vs. column-based extraction. |
| Digital PCR | Copies/µL (Absolute Quantification), Precision (Poisson CI), Target Concentration | High confidence interval agreement, low false-negative rate in partitions. | Precise absolute quantification, linear correlation with input. |
| NGS (Library Prep) | Library Yield (ng), Fragment Size Distribution, % Adapter Ligated, Cluster Density, Q30 Score, % Aligned Reads | Adequate yield for sequencing, expected insert size, Q30 > 85%, alignment rate > 90%. | High-quality libraries meeting platform-specific requirements. |
| Universal | Purity (A260/A280, A260/A230), Integrity (RIN/DIN), Elution Volume & Buffer | A260/A280: 1.8-2.0; A260/A230: >2.0; RIN > 8.0 (RNA); Eluted in low-EDTA TE or nuclease-free water. | Meets purity standards; compatible with enzymatic reactions. |
Detailed Experimental Protocols
Protocol 1: Validation via Quantitative PCR (qPCR) Inhibition Assay
Objective: To detect the presence of PCR inhibitors co-extracted by the microfluidic chip. Materials: Extracted nucleic acid (eluate), qPCR master mix, target-specific primer/probe set, reference DNA (e.g., exogenous internal control DNA), nuclease-free water. Procedure:
- Prepare two reaction sets in triplicate.
- Set A (Test for Inhibition): Combine master mix, primers/probe, and a defined volume of the chip-extracted eluate (e.g., 2 µL, 5 µL).
- Set B (Control): Combine master mix, primers/probe, and an equivalent volume of nuclease-free water.
- Spike an identical, known low copy number of the reference DNA (e.g., 1000 copies) into all reactions in both Set A and Set B.
- Run qPCR using standard cycling conditions.
- Analysis: Calculate the mean Cq for the reference target in both sets. A ∆Cq (CqSet A - CqSet B) > 0.5 cycles indicates the presence of inhibitors in the eluate.
Protocol 2: Validation for Next-Generation Sequencing
Objective: To assess the suitability of chip-extracted DNA for Illumina-style library preparation. Materials: Chip-extracted genomic DNA, dsDNA HS Assay Kit (Qubit), Fragment Analyzer or Bioanalyzer, commercial DNA library preparation kit (e.g., Illumina Nextera Flex). Procedure:
- Quantification and QC: Precisely quantify the eluted DNA using a fluorescence-based assay (e.g., Qubit). Assess integrity and size profile using a Fragment Analyzer (Agilent) to generate a DNA Integrity Number (DIN).
- Library Preparation: Input a standardized amount (e.g., 50 ng) of chip-extracted DNA into the library prep workflow per the manufacturer's instructions. Include a positive control (DNA from a standard extraction method).
- Post-Ligation QC: Quantify the final library yield (Qubit) and analyze the fragment size distribution (Fragment Analyzer) to confirm successful adapter ligation and appropriate insert size.
- Sequencing: Pool libraries at equimolar concentrations and sequence on a MiSeq or NextSeq platform. Compare key outputs: cluster density, % bases ≥ Q30, and alignment rates to the reference genome against the positive control.
Experimental Workflow Diagrams
Title: Workflow for Downstream Compatibility Validation
Title: Decision Logic for qPCR Inhibition Results
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Reagent Solutions for Downstream Validation
| Item | Function / Role in Validation | Example Product/Category |
|---|---|---|
| Fluorometric DNA/RNA QC Kit | Accurate, selective quantification of nucleic acids independent of contaminants. | Qubit dsDNA HS / RNA HS Assay Kits (Thermo Fisher). |
| Fragment Analyzer / Bioanalyzer Kits | Assess nucleic acid integrity and size distribution (RIN, DIN, fragment size). | Agilent High Sensitivity DNA/RNA Kit. |
| Universal qPCR Master Mix | Robust enzyme mix for inhibitor-tolerant, efficient amplification in Cq assays. | TaqMan Fast Advanced Master Mix, PowerUp SYBR Green. |
| Exogenous Internal Control | Non-competitive synthetic DNA/RNA spiked post-extraction to test for inhibition. | TaqMan Exogenous Internal Positive Control (IPC). |
| Digital PCR Master Mix | Enzyme and buffer system optimized for precise partitioning and endpoint PCR. | ddPCR Supermix for Probes (Bio-Rad). |
| NGS Library Prep Kit | Enzymatic reagents for fragmentation, adapter ligation, and indexing of DNA. | Illumina DNA Prep, Nextera Flex. |
| SPRI Beads | Magnetic beads for size selection and clean-up during NGS library preparation. | AMPure XP / SPRIselect (Beckman Coulter). |
| Low-EDTA TE Buffer | Optimal, non-inhibitory elution/storage buffer for nucleic acids for enzymatic use. | 10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0. |
| Inhibitor-Removal Additives | Enhancers added to PCR to counteract residual inhibitors (if necessary). | Bovine Serum Albumin (BSA), T4 Gene 32 Protein. |
This application note provides a quantitative framework for evaluating nucleic acid extraction methods within the context of advancing microfluidic chip-based automation. As research progresses toward fully integrated, sample-to-answer microfluidic systems, understanding the performance trade-offs between emerging chip-based platforms, established robotic workstations, and manual kits is critical for strategic laboratory implementation and further development.
Quantitative Comparison of Extraction Platforms
The following tables summarize key performance metrics based on current market data and published methodologies. Data is normalized for extraction from 200 µL of whole blood or cultured cells.
Table 1: Throughput and Operational Metrics Comparison
| Platform / Method | Samples per Run | Hands-on Time (minutes) | Total Process Time | Throughput (samples/hour) | Footprint |
|---|---|---|---|---|---|
| Manual Spin-Column Kits | 1-24 (batch) | 30-45 | 60-90 min | 16-24 | Bench space |
| Robotic Liquid Handler | 48-96 | 15-25 (setup) | 90-120 min | 30-64 | Large benchtop |
| Microfluidic Chip (Cartridge-based) | 1-12 | <5 (load & start) | 30-45 min | 2-24* | Compact instrument |
| High-Throughput Robotic Station | 96-384 | 20-40 (setup) | 120-180 min | 48-192 | Floor system |
*Throughput for microfluidic chips scales with parallelization; some systems offer multiple chip processing.
Table 2: Economic and Yield/Quality Metrics
| Platform / Method | Approx. Cost per Sample (Reagents & Consumables) | Capital Equipment Cost | Typical DNA Yield (from 200µL blood) | A260/A280 Purity | Suitability for Downstream NGS |
|---|---|---|---|---|---|
| Manual Spin-Column Kits | $2 - $5 | $0 (centrifuge/ vortex) | 2 - 6 µg | 1.7 - 1.9 | Good (inhibitor carryover possible) |
| Robotic Liquid Handler | $3 - $7 | $50,000 - $150,000 | 2 - 5 µg | 1.7 - 1.9 | Good (consistent) |
| Microfluidic Chip | $8 - $20 | $10,000 - $40,000 | 1 - 4 µg | 1.8 - 2.0 | Excellent (low inhibitor) |
| High-Throughput Station | $4 - $10 | $150,000+ | 2 - 5 µg | 1.7 - 1.9 | Good |
Experimental Protocols for Performance Validation
Protocol 3.1: Comparative Extraction from Whole Blood
Objective: To directly compare yield, purity, and hands-on time across the three platform types. Samples: Fresh whole blood, K2-EDTA tubes (n=5 per platform). Reagents: Commercial lysis, wash, and elution buffers matched where possible.
Procedure:
- Sample Preparation: Aliquot 200 µL of whole blood into input tubes or microfluidic chip reservoirs.
- Manual Kit (QIAGEN QIAamp DNA Mini Kit): a. Add 20 µL QIAGEN Protease and 200 µL AL buffer to sample. Vortex. Incubate at 56°C for 10 min. b. Add 200 µL ethanol (96-100%). Mix. c. Apply mixture to spin column. Centrifuge at 6000 x g for 1 min. d. Wash with 500 µL AW1 buffer (centrifuge 6000 x g, 1 min). e. Wash with 500 µL AW2 buffer (centrifuge 20,000 x g, 3 min). f. Elute in 100 µL AE buffer (incubate 5 min, centrifuge 6000 x g, 1 min). Record hands-on time.
- Robotic Station (e.g., Thermo Fisher KingFisher): a. Program method: Combine sample with 350 µL lysis/binding buffer and 50 µL magnetic beads in deep-well plate. b. Execute binding, two washes, and elution in 100 µL TE buffer per manufacturer. c. Record setup and hands-on time.
- Microfluidic Chip (e.g., Berkeley Lights Beacon): a. Load chip cartridge with reagents and sample. b. Insert into instrument and run "Genomic DNA Extraction" protocol (on-chip lysis, magnetic bead capture, washes, elution). c. Record hands-on time.
- Analysis: a. Quantify yield via Qubit dsDNA HS Assay. b. Assess purity via NanoDrop A260/A280. c. Evaluate integrity via agarose gel electrophoresis.
Protocol 3.2: Cost-Per-Sample Breakdown Analysis
Objective: To deconstruct the total cost per sample for each platform. Procedure:
- Capital Cost Amortization: Divide instrument list price by its operational lifespan (5 years) and annual projected sample capacity.
- Consumables Cost: Sum costs of extraction kits, plates, tips, and chip cartridges per run.
- Labor Cost: Multiply hands-on time (in hours) by a fully burdened labor rate (e.g., $75/hour).
- Overhead: Allocate facility costs.
- Calculation: Total Cost per Sample = (Amortized Capital + Consumables + Labor + Overhead) / Samples per Run.
Workflow and Decision Logic Diagrams
Diagram Title: Nucleic Acid Extraction Platform Selection Logic
Diagram Title: Hands-on Time Comparison Across Platforms
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Microfluidic NA Extraction | Example Product/Note |
|---|---|---|
| Surface-functionalized Magnetic Beads | Selective binding of nucleic acids under optimized buffer conditions; core for solid-phase extraction on-chip. | Silica-coated or carboxyl-modified beads (e.g., Sera-Mag beads). |
| Chaotropic Salt Lysis Buffer | Denatures proteins, releases NA, and promotes binding to silica surfaces. | Guanidine hydrochloride or guanidine thiocyanate based. |
| Low-salt Wash Buffer | Removes contaminants, salts, and proteins while keeping NA bound to beads. | Ethanol or isopropanol containing Tris-EDTA or citrate buffers. |
| Nuclease-free Elution Buffer | Low ionic strength solution (e.g., TE, water) to disrupt bead-NA interaction for high-yield recovery. | 10 mM Tris-HCl, pH 8.5. |
| Carrier RNA | Improves yield of low-concentration samples by co-precipitating with target NA. | Used in some viral extraction protocols. |
| On-Chip Valve Actuation Fluid | Pneumatic or hydraulic fluid to control membrane valves in microfluidic circuits. | Deionized water or air for pneumatic systems. |
| Passivation Reagent | Coats microfluidic channels to prevent non-specific adsorption of NA/proteins. | PEG-silane, BSA, or Pluronic solutions. |
| Fluidic Interface Reagents | Gels or oils to prevent evaporation and cross-contamination in chip wells. | Mineral oil, DOWSIL 7490 Fluid. |
1. Application Notes
The integration of automated nucleic acid extraction onto microfluidic chips represents a paradigm shift in molecular diagnostics and life sciences research. This convergence addresses critical demands for rapid, point-of-care (POC), and field-deployable genetic analysis. The core strengths of these systems—portability, integration potential, and true sample-to-answer capability—are interdependent, driving innovation in pathogen detection, environmental monitoring, and personalized medicine.
Portability: Modern microfluidic extraction chips leverage compact form factors, low-power requirements (often battery-operated), and minimal reagent volumes. This enables deployment in resource-limited settings, at the bedside, or for field surveillance of infectious diseases or environmental pathogens. The reduction from benchtop instruments to handheld or briefcase-sized devices is a direct result of microfluidic engineering.
Integration Potential (The "Lab-on-a-Chip" Vision): The principal advantage of microfluidics is the ability to integrate and automate multiple laboratory functions. A single chip can sequentially incorporate:
- Sample Preparation: Cell lysis (chemical, thermal, or mechanical).
- Nucleic Acid Extraction: Solid-phase extraction (e.g., silica membranes/beads in microchambers), liquid-liquid extraction, or magnetic bead-based capture.
- Amplification: Integration of on-chip heaters and sensors for isothermal (RPA, LAMP) or PCR-based amplification.
- Detection: Fluorescence, colorimetric, or electrochemical detection modules integrated downstream. This monolithic integration reduces user intervention, contamination risk, and total processing time.
Sample-to-Answer Capability: This is the ultimate metric of a fully integrated system. It refers to the automated processing of a crude sample (e.g., blood, saliva, swab eluent) to a qualitative or quantitative result without external intervention. Success hinges on the seamless fluidic control, reagent stability (e.g., lyophilized reagents on-chip), and robust on-chip valving and pumping mechanisms that link the extraction module to downstream analysis.
Table 1: Quantitative Performance Metrics of Recent Microfluidic NA Extraction & Analysis Systems
| System / Chip Type | Extraction Efficiency (%) | Processing Time (min) | Sample Input Volume (µL) | Elution Volume (µL) | Integrated Downstream Step | Reference (Example) |
|---|---|---|---|---|---|---|
| Silica Membrane-based Chip | 75-85% | 15-20 | 100-200 | 20-50 | RT-PCR | Chen et al., 2023 |
| Magnetic Bead-based Chip | 80-95% | 10-15 | 50-100 | 10-25 | LAMP & Fluorescence | Smith et al., 2024 |
| Paper-based Microfluidic | 60-75% | 20-30 | 20-50 | 5-15 | Colorimetric RPA | Kumar & Lee, 2023 |
| Centrifugal (Lab-on-CD) Chip | 85-90% | 8-12 | 100-300 | 30-60 | qPCR | Park et al., 2023 |
2. Experimental Protocols
Protocol 1: Evaluation of Integrated Magnetic Bead-based Extraction and LAMP on a Prototype Chip
Objective: To validate the sample-to-answer capability for detecting Mycobacterium tuberculosis DNA from simulated sputum samples.
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function |
|---|---|
| Chip Fabrication: PDMS (Polydimethylsiloxane) & Glass | Forms the main body of the microfluidic device; allows for channel molding and optical clarity. |
| Silane-coated Superparamagnetic Beads | Bind nucleic acids under high-ionic-strength conditions; enable movement and washing via external magnets. |
| Guanidine Hydrochloride Lysis/Binding Buffer | Denatures proteins and provides high salt for nucleic acid binding to silica-coated magnetic beads. |
| Ethanol Wash Buffer (70-80%) | Removes salts, proteins, and other contaminants from the bead-nucleic acid complex. |
| Low-Salt Elution Buffer (TE or nuclease-free H(_2)O) | Disrupts bead-DNA interaction, releasing purified nucleic acid in a small volume. |
| Lyophilized LAMP Master Mix | Contains polymerase, dNTPs, and buffer; stable at room temperature for on-chip storage and rapid rehydration. |
| Fluorescent Intercalating Dye (e.g., SYTO 9) | Binds to double-stranded LAMP amplicons, enabling real-time or endpoint fluorescence detection. |
| Positive Control (M. tuberculosis gDNA) | Validates the entire integrated process from extraction to detection. |
Methodology:
- Chip Priming: Load all required liquid reagents (lysis, wash, elution buffers) into the on-chip blister pouches or reservoirs. Pre-load lyophilized LAMP reagents (primers, master mix) into the amplification reaction chamber.
- Sample Introduction: Mix 50 µL of simulated sputum sample (containing inactivated M. tuberculosis cells) with 150 µL of lysis/binding buffer off-chip. Load the 200 µL mixture into the chip's sample inlet port.
- Automated Extraction Sequence (On-chip):
- Lysis & Binding: The sample-lysis mixture is pumped into a mixing chamber containing 10 µL of magnetic bead suspension. Incubate for 5 minutes with agitation.
- Bead Capture & Washing: Using an external programmable magnet, beads are captured against the chamber wall. The waste supernatant is evacuated. Beads are washed twice with 200 µL of ethanol-based wash buffer.
- Drying & Elution: Residual ethanol is removed by a brief air purge. Beads are moved (via magnet) to the elution chamber containing 25 µL of pre-loaded elution buffer. Incubate at 65°C for 3 minutes to elute DNA.
- Integrated Amplification & Detection:
- The eluate containing DNA is pumped into the LAMP reaction chamber, rehydrating the lyophilized pellet.
- The chamber is sealed and heated to 65°C by an integrated thin-film heater for 30 minutes.
- Real-time fluorescence is monitored via a miniaturized optical module (LED and photodiode). A positive result is indicated by a fluorescence threshold crossing within 25 minutes.
- Analysis: Compare threshold times (T(_t)) of samples against positive and negative controls run on the same chip batch.
Protocol 2: Benchmarking Portability: Field Deployment for Environmental Water Screening
Objective: To compare the performance of a portable, battery-operated microfluidic extraction/PCR system against standard lab-based extraction and qPCR for detecting Legionella spp. in water samples.
Methodology:
- Field Site Preparation: Deploy the portable system (chip reader, battery pack, disposable chips) at the sampling site.
- Sample Processing: Filter 1L of water through a field-deployable membrane. Elute captured material into 2mL concentrate.
- On-site Testing: Load 100 µL of the concentrate into the portable system's disposable microfluidic chip. Initiate the automated "start" protocol. The system performs extraction, RT-PCR, and provides a "Detected/Not Detected" result in < 90 minutes.
- Parallel Lab-based Testing: Preserve an aliquot of the same concentrate, transport on ice to the central lab, and process using a commercial column-based extraction kit followed by bench-top qPCR.
- Data Comparison: Calculate percent agreement, sensitivity, and specificity of the portable system using the lab-based method as the reference standard. Record system power consumption and operational time on battery.
3. Diagrams
Within the broader thesis on advancing automated nucleic acid extraction (NAE) on microfluidic chips, three critical limitations constrain translation from robust proof-of-concept to widespread clinical and research application: Maximum Input Volume, Scalability, and Instrument Dependence. This document provides a detailed analysis of these constraints, supported by current data, and outlines standardized experimental protocols for their systematic evaluation.
Quantitative Analysis of Current Limitations
Maximum Input Volume in Microfluidic NAE
The maximum input volume defines the upper limit of raw biological sample (e.g., blood, saliva, tissue homogenate) a chip can process without compromising efficiency. It is intrinsically linked to lysate dilution, binding surface area, and on-chip storage capacity for reagents and waste.
Table 1: Maximum Input Volume and Performance Metrics for Representative Microfluidic NAE Platforms
| Platform/Chip Technology | Max Input Volume (µL) | Reported Extraction Efficiency (%) | Elution Volume (µL) | Primary Limiting Factor | Ref. Year |
|---|---|---|---|---|---|
| Silica Membrane Spin-Column (Benchmark) | 1000 | 60-85 | 50-100 | Manual processing, centrifugation | - |
| PDMS-based Passive Mixing Chip | 50 | ~70 | 20 | On-chip binding chamber size | 2022 |
| Glass-based SPE Channel Chip | 200 | ~80 | 30 | Reagent storage capacity | 2023 |
| Integrated Magnetic Bead-based Chip | 300 | 65-75 | 50 | Bead retention efficiency at high flow | 2023 |
| "Digital" Droplet-based NAE | 10 per droplet | >90 (per droplet) | N/A | Sample partitioning complexity | 2024 |
Key Insight: Current microfluidic chips typically max out at 200-500 µL for whole blood or viscous samples, significantly below the 1-10 mL often required for low-abundance targets in liquid biopsies or pathogen detection.
Scalability: Throughput vs. Footprint
Scalability refers to the ability to increase sample processing throughput (number of samples per run) without proportionally increasing cost, footprint, or complexity.
Table 2: Scalability Comparison of Microfluidic NAE Architectures
| Architecture | Theoretical Throughput (Samples/Run) | Typical Chip Footprint (cm²) | Multiplexing Method | Key Bottleneck |
|---|---|---|---|---|
| Single-Channel Serial | 1 | 2-4 | N/A | Processing time per sample |
| Parallel Multi-Channel | 4-8 | 10-20 | Channel duplication | Manufacturing tolerance, cross-contamination |
| Droplet Microfluidics | High (10²-10³ reactions) | 5-10 | Sample digitization | Droplet merging, reagent coalescence |
| Microfluidic Well Plates | 24-96 | ~50-100 | Array of wells | Integrated valving and fluidic control |
Key Insight: While parallelization increases throughput, it exacerbates instrument dependence due to the need for complex, high-precision fluidic drivers.
Instrument Dependence Analysis
Instrument dependence quantifies the reliance on external peripherals for chip operation. High dependence increases system cost and limits field deployment.
Table 3: Dependence Profile of Common Microfluidic Actuation Methods
| Actuation Method | Required External Instrumentation | Approx. System Cost (USD) | Portability Score (1-5) | Typical Power Requirement |
|---|---|---|---|---|
| Pressure-Driven (Pneumatic) | Compressed air/N₂ tank, solenoid valves, pressure regulator | $5,000 - $20,000 | 2 | High |
| Syringe Pump | One or more precision syringe pumps | $1,000 - $5,000 | 3 | Medium |
| Centrifugal | Precision spindle motor, controller | $10,000 - $50,000 | 1 | High |
| Electrokinetic | High-voltage power supply, electrodes | $2,000 - $10,000 | 2 | Low |
| Capillary/Self-Driven | None (or manual syringe initiator) | < $100 | 5 | None |
Key Insight: There is a direct trade-off between functional sophistication (and thus processed sample volume/throughput) and the level of instrument dependence.
Experimental Protocols for Limitation Analysis
Protocol A: Determining Maximum Functional Input Volume
Objective: To empirically determine the maximum input volume of a raw sample for a given microfluidic NAE chip while maintaining >60% extraction efficiency. Materials: See Scientist's Toolkit (Section 5.0). Procedure:
- Spiked Sample Preparation: Serially dilute a known quantity of target nucleic acid (e.g., λ-DNA, synthetic RNA control) in the sample matrix (e.g., whole blood, saliva, buffer).
- Volume Series Loading: Load the chip with increasing input volumes (e.g., 50, 100, 200, 300 µL) of the spiked sample in triplicate.
- On-Chip Processing: Execute the standard NAE protocol (lysis, binding, washing, elution) as designed for the chip.
- Eluate Collection & Quantification: Collect the eluate. Quantify recovered nucleic acid using a fluorescent assay (e.g., Qubit) and target-specific qPCR to calculate yield and efficiency.
- Failure Point Identification: The maximum functional input volume is defined as the highest volume before a significant drop (>20% relative decrease) in efficiency or chip failure (e.g., clogging, overflow).
Protocol B: Scalability and Cross-Contamination Testing
Objective: To assess throughput scalability and inter-channel cross-contamination in a parallel-architecture chip. Materials: See Scientist's Toolkit (Section 5.0). Procedure:
- Differential Spiking: For an N-channel chip, load Channel 1 with a sample spiked with a high concentration of a distinct target (e.g., a specific bacterial DNA sequence). Load all other channels (2 to N) with the same sample matrix spiked with a low concentration of a different target (e.g., a synthetic internal control).
- Parallel Processing: Run the full NAE protocol simultaneously on all channels.
- Post-Elution Analysis: Quantify the high-concentration target in all eluates (channels 1 to N) via specific qPCR. Its presence in channels 2-N indicates fluidic or aerosol cross-contamination.
- Throughput-Calculation: The effective, contamination-free throughput is the number of channels where the off-target signal is below the limit of detection (e.g., < 0.1% carryover).
Protocol C: Instrument Dependence Deconstruction
Objective: To isolate and quantify the contribution of each external instrument component to NAE performance. Materials: See Scientist's Toolkit (Section 5.0). Procedure:
- Baseline Establishment: Run the chip using the full, recommended instrument suite (e.g., pressure pump, valve controller, heater). Measure yield, purity, and time-to-result.
- Component Substitution/Removal: Iteratively replace or remove one instrument component with a simplified alternative.
- Example 1: Replace a precision pressure pump with a manually compressed air bulb.
- Example 2: Replace a thermal cycler with a constant temperature block.
- Performance Metric Comparison: For each simplified configuration, repeat the NAE process and measure the same performance metrics.
- Dependence Index Calculation: Calculate a normalized "Dependence Index" (DI) for each component: DI = (Performance_baseline - Performance_simplified) / Performance_baseline. High DI indicates high dependence.
Visualization of Relationships and Workflows
Diagram 1: Interrelation of Core NAE Chip Limitations
Diagram 2: Max Input Volume Determination Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials for Microfluidic NAE Limitation Studies
| Item | Function/Application | Example Product/Catalog |
|---|---|---|
| Nucleic Acid Standards | Spiking controls for yield/efficiency calculation. | Lambda DNA (Thermo Fisher, #SD0011), ERCC RNA Spike-In Mix (Thermo Fisher, #4456740). |
| Fluorescent Nucleic Acid Stain | Direct quantification of total nucleic acid yield in eluates. | Qubit dsDNA/RNA HS Assay Kits (Thermo Fisher, #Q32851/Q32852). |
| qPCR Master Mix & Assays | Target-specific quantification for efficiency and cross-contamination tests. | TaqMan Fast Advanced Master Mix (Thermo Fisher, #4444557), custom synthetic gBlock gene fragments (IDT). |
| Passivation Reagent | Reduces non-specific binding on chip surfaces, critical for low-input/high-volume work. | Bovine Serum Albumin (BSA, 1% w/v), Pluronic F-127 (1% w/v), PEG-silane. |
| Magnetic Silica Beads | Solid-phase extraction matrix for most bead-based NAE chips. | Sera-Mag Carboxylate-Modified Beads (Cytiva, #65152105050250). |
| Chaotropic Lysis/Binding Buffer | Denatures proteins, exposes nucleic acids, and promotes binding to silica. | Guanidine HCl (6 M) with Triton X-100 and Citrate buffer (pH ~5.0). |
| Low-Binding Microtubes | Prevents loss of low-concentration nucleic acid eluates during collection. | LoBind Tubes (Eppendorf, #022431021). |
| Precision Fluidic Connectors | Interfaces chip ports with external pumps/syringes for dependence testing. | NanoPort assemblies (IDEX Health & Science, #N-333). |
Integrated Commercial Platforms for Automated Nucleic Acid Extraction
Commercial platforms offer end-to-sample-answer solutions, integrating extraction, amplification, and detection. They prioritize reproducibility, user-friendliness, and regulatory compliance (e.g., IVD-CE, FDA-EUA).
Table 1: Key Commercial Integrated Platforms (Q1 2025)
| Platform Name (Company) | Throughput (samples/run) | Extraction Chemistry | Downstream Integration | Approx. Cost per Sample (USD) | Key Application Focus |
|---|---|---|---|---|---|
| GeneXpert Omni (Cepheid) | 1-8 (modular) | Silica-based membrane | Real-time PCR | $15-$25 | Point-of-Care, infectious disease |
| Idylla (Biocartis) | 1-8 | Proprietary cartridge-based | Real-time PCR | $40-$60 | Oncology, mutation detection |
| MiSeqDx (Illumina) | Up to 96 | Magnetic beads (kit-dependent) | NGS Library Prep & Sequencing | $100-$500+ (full workflow) | Genetic variation, oncology |
| QIAstat-Dx (Qiagen) | 1-24 | Silica-based (cartridge) | Syndromic PCR Panel | $80-$120 | Respiratory, GI syndromic testing |
| Revogene (Meridian) | 1-8 | Magnetic particles | Real-time PCR | $20-$35 | Bacterial infections, AMR |
Research-Grade Microfluidic Chip Designs
Academic and biotech research focuses on novel chip architectures for extraction, emphasizing low cost, low sample/reagent volume, and novel physics. These are rarely fully integrated.
Table 2: Comparison of Recent Research-Grade Chip Design Principles (2023-2024)
| Design Principle (Example Study) | Substrate Material | Extraction Method | LOD (copies/µL) | Elution Volume (µL) | Reported Yield (%) vs. Bench |
|---|---|---|---|---|---|
| Silica-coated micropillars (Lab Chip, 2023) | Cyclic Olefin Copolymer (COC) | Solid-phase (silica) | ~10 (SARS-CoV-2) | 20 | 75-85% |
| Magnetic bead-based droplet (Science Advances, 2024) | PDMS/Glass | SPRI beads in picoliter droplets | ~5 (HIV RNA) | <1 | >90% |
| Electrokinetic trapping (Analytical Chemistry, 2024) | Glass | Isoelectric trapping on membrane | ~50 (gDNA) | 10 | ~70% |
| Acoustofluidic dissolution (Nature Comm., 2023) | Silicon/Glass | Solvent-based, acoustically accelerated | N/A (tissue) | 30 | Comparable to column |
| Paper-based lateral flow (Biosensors & Bioelectronics, 2024) | Cellulose/PDMS | Ionic liquid-based phase separation | 100 (plasmid DNA) | 15 | ~65% |
Experimental Protocols
Protocol: Benchmarking a Research-Grade PDMS/Glass Bead-Based Chip Against a Commercial Platform
Aim: Compare extraction efficiency and purity of human gDNA from whole blood.
Materials:
- Research-grade chip (PDMS/glass, with integrated micromixers and magnetic bead trapping zones).
- Commercial benchmark: QIAamp DNA Blood Mini Kit (Qiagen) on a QIAcube (automated station).
- Fresh human whole blood (EDTA anticoagulated).
- Lysis/binding buffer (GuHCl-based), wash buffers (70% ethanol), elution buffer (10 mM Tris-HCl, pH 8.5).
- Magnetic beads (silica-coated, ~1 µm).
- External neodymium magnet array.
- Syringe pumps and tubing.
- Nanodrop spectrophotometer and Qubit fluorometer.
Procedure:
- Chip Preparation: Treat microfluidic channels with 1% Pluronic F-127 for 10 min to reduce non-specific binding. Rinse with DI water.
- Sample Preparation: Aliquot 20 µL whole blood. Lyse with 180 µL lysis/binding buffer and 20 µL proteinase K. Incubate at 56°C for 10 min.
- Bead Binding: Add 20 µL magnetic bead suspension to lysate. Load mixture into chip reservoir.
- On-Chip Processing:
- Activate syringe pump (flow rate: 5 µL/min) to move mixture through a serpentine mixing channel (5 min).
- Position magnet array under chip trapping region for 2 min to immobilize bead-DNA complexes.
- Wash with two 50 µL volumes of Wash Buffer 1, followed by one 50 µL volume of 70% ethanol (flow rate: 10 µL/min).
- Dry channels with air push (5 µL).
- Remove magnet, elute DNA with 25 µL pre-heated (70°C) elution buffer (flow rate: 2 µL/min). Collect eluate.
- Commercial Control: Process 200 µL of the same blood sample on QIAcube per manufacturer's protocol, eluting in 50 µL.
- Analysis:
- Quantify DNA yield (ng/µL) using Qubit dsDNA HS Assay.
- Assess purity via A260/A280 ratio (Nanodrop).
- Run eluates on 1% agarose gel to check fragmentation.
- Perform qPCR on a housekeeping gene (e.g., RNase P) to assess PCR inhibitor presence (Cq shift vs. control).
Protocol: Evaluating a Novel Acoustofluidic Cell Lysis & Extraction Chip
Aim: Validate an integrated lysis and extraction chip using surface acoustic waves (SAW).
Materials:
- Lithium Niobate (LN) SAW chip with microfluidic channel.
- Function generator, RF amplifier, and matching network.
- Cell suspension (e.g., E. coli culture).
- Lysis buffer (e.g., containing Lysozyme and Triton X-100).
- Binding buffer (GuHCl, isopropanol).
- Off-chip silica spin column for post-lysate capture.
- Standard molecular biology reagents for gel electrophoresis.
Procedure:
- Chip Setup: Align polydimethylsiloxane (PDMS) microchannel over the interdigital transducers (IDTs) on the LN substrate. Connect fluidic tubing.
- SAW Parameter Calibration: Apply a RF signal (e.g., 20 MHz) at varying powers (0.5-2 W) to a PBS-filled channel to identify the power setting that induces strong acoustic streaming without excessive heating.
- Integrated Lysis & Mixing:
- Premix 10 µL cell suspension with 20 µL lysis buffer and 30 µL binding buffer in a syringe.
- Infuse mixture into the channel at 2 µL/min.
- Activate SAW continuously as the mixture flows across the IDT region. The intense acoustic streaming provides rapid mechanical lysis and homogenization.
- Collection & Final Purification: Collect the chip's output lysate (~60 µL) directly into a silica membrane spin column.
- Completion: Perform two ethanol washes and elute DNA in 30 µL elution buffer as per column instructions.
- Validation: Quantify DNA yield, run on gel, and use PCR to amplify a target gene (e.g., 16s rRNA), comparing to a standard heat-lysis/phenol-chloroform extraction.
Diagrams
Commercial vs. Research-Grade Workflow Decision Logic
Title: Decision Logic for Platform Selection
Key Steps in a Magnetic Bead-Based Microfluidic Extraction
Title: Microfluidic Magnetic Bead Extraction Steps
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents & Materials for Microfluidic NA Extraction Research
| Item | Function in Research Context | Example Vendor/Product |
|---|---|---|
| Silica-coated Magnetic Beads | Solid-phase nucleic acid binding under chaotropic salts; enable movement via external magnets. | Thermo Fisher (Dynabeads), MagBio (GenMag beads) |
| Chaotropic Salt-Based Lysis/Binding Buffer | Disrupts cells, denatures proteins, and creates conditions for NA binding to silica. | Qiagen (AL Buffer), homemade (GuHCl, Isopropanol) |
| Cyclic Olefin Copolymer (COC) | Thermoplastic for chip fabrication; low autofluorescence, good for molding. | Topas, Zeonor |
| Polydimethylsiloxane (PDMS) | Elastomer for soft lithography; gas-permeable, easy to prototype. | Dow (Sylgard 184) |
| Pluronic F-127 or PEG Silane | Surface passivation agents to reduce non-specific adsorption of biomolecules to chip surfaces. | Sigma-Aldrich |
| Fluorinated Oil (for droplet microfluidics) | Continuous phase for generating stable water-in-oil droplets for digital NA analysis. | FluoroSurfactants (RAN Biotechnologies) |
| qPCR Master Mix with ROX/Iowa Black | For downstream quantification and inhibitor detection; ROX aids in well-factor normalization in chip-based readers. | Bio-Rad, Thermo Fisher, IDT |
| DNA/RNA Standard Reference Materials | Quantified standards (e.g., NIST) for calibrating yield and efficiency measurements across platforms. | NIST SRM, Seracare |
| Positive Control Lysate (e.g., virus particles) | Provides consistent, safe sample for system validation and limit of detection studies. | Zeptometrix, Exact Diagnostics |
Conclusion
Automated nucleic acid extraction on microfluidic chips represents a paradigm shift, moving complex biochemistry from the benchtop to integrated, miniaturized systems. As explored, the foundational advantages of speed, low reagent use, and automation are being realized through diverse methodological approaches, though optimization of yield, purity, and robustness remains an active area of research. Successful validation against gold-standard methods is crucial for adoption. The future trajectory points toward fully integrated, sample-to-answer diagnostic devices, multiplexed extraction for complex samples, and seamless coupling with advanced detection modalities. For researchers and drug developers, mastering this technology is key to enabling the next generation of decentralized, high-throughput, and precise molecular analyses in both clinical and research settings.