Non-Viral Nanoparticle Vectors for Gene Therapy: Current Advances, Clinical Applications, and Future Directions
This article provides a comprehensive analysis of non-viral nanoparticle vectors for gene therapy, a field gaining significant momentum as a safer and more scalable alternative to viral vectors.
Non-Viral Nanoparticle Vectors for Gene Therapy: Current Advances, Clinical Applications, and Future Directions
Abstract
This article provides a comprehensive analysis of non-viral nanoparticle vectors for gene therapy, a field gaining significant momentum as a safer and more scalable alternative to viral vectors. Tailored for researchers, scientists, and drug development professionals, it explores the foundational principles of lipid-based, polymer-based, and inorganic nanoparticles. The scope extends to methodological advances in delivering complex cargos like CRISPR/Cas9, troubleshooting of key challenges such as transfection efficiency and tissue-specific targeting, and a critical validation against established viral platforms. By synthesizing the latest research and clinical progress, this review serves as a strategic resource for navigating the development and application of non-viral gene delivery technologies.
The Rise of Non-Viral Vectors: Overcoming the Limitations of Viral Gene Delivery
Viral vectors, including adeno-associated virus (AAV), lentivirus, and adenovirus, are foundational tools in modern gene therapy and biological research, enabling efficient gene delivery both in vivo and in vitro [1]. Their ability to provide high transduction efficiency and long-term transgene expression has supported the development of approved treatments for conditions such as spinal muscular atrophy, inherited retinal dystrophy, and β-thalassemia [1]. However, their clinical application is constrained by significant challenges related to manufacturing scalability, immunogenicity, and insertional mutagenesis risks [2] [3]. These limitations underscore the clinical imperative to develop robust solutions that enhance the safety, efficiency, and scalability of viral vector technologies. This application note details these challenges and presents optimized protocols to address them, providing researchers with actionable methods to improve viral vector performance in critical experiments.
Current Challenges in Viral Vector Applications
The clinical and research use of viral vectors faces several persistent hurdles that impact the efficacy, safety, and practicality of gene delivery systems.
Manufacturing and Characterization Bottlenecks: The transition of gene therapies from clinical development to commercial licensure demands a substantial increase in viral vector manufacturing capacity—estimated at 1–2 orders of magnitude for many promising disease indications [2]. This scaling challenge is compounded by the need for rigorous characterization of critical quality attributes. Current analytical techniques for assessing attributes such as empty/full capsid ratios, titer, and post-translational modifications often suffer from low throughput, large sample requirements, and poorly understood measurement variability [4].
Safety and Immunogenicity Concerns: Retroviral and lentiviral vectors pose risks of insertional mutagenesis, where integration into the host genome can disrupt or dysregulate genes, potentially leading to oncogenic transformation [3]. Although improved self-inactivating (SIN) designs have reduced this risk, monitoring remains crucial [2] [3]. Immune responses also present barriers; for instance, AAV therapies can trigger reactions that limit transgene expression or necessitate immunosuppression [1].
Technical Limitations in Complex Systems: Efficient gene delivery remains challenging in physiologically relevant 3D models such as organoids. Their complex architecture presents significant barriers to uniform transduction, limiting their utility in assessing vector performance and dose-response relationships [5] [6].
Table 1: Key Challenges and Current Limitations in Viral Vector Applications
| Challenge Category | Specific Limitation | Impact on Research/Therapy |
|---|---|---|
| Manufacturing & Scalability | Limited production capacity for commercial-scale supply [2] | Restricts patient access and increases costs |
| Product Characterization | Lack of high-throughput, precise analytical methods [4] | Hampers quality control and lot consistency |
| Delivery Efficiency | Low transduction efficiency in 3D organoid systems [5] | Reduces predictive value in preclinical models |
| Safety & Monitoring | Risk of insertional mutagenesis with integrating vectors [3] | Requires long-term patient monitoring (up to 15 years) |
| Immune Response | Pre-existing or therapy-induced immunity to viral capsids [1] | Limits transduction efficiency and re-dosing potential |
Quantitative Analysis of Viral Vector Performance
Recent meta-analytic data highlights the variable protective efficacy of different viral vector platforms. A 2025 systematic review and meta-analysis of vaccine strategies for foot-and-mouth disease virus (FMDV)—a model for viral vector research—demonstrated clear efficacy differences between platforms [7]. Subgroup analysis revealed that VLP and viral vector vaccines offered higher protection rates compared to other platforms, though wide confidence intervals indicate significant variability across studies [7]. This heterogeneity underscores the influence of vector design and production methods on clinical outcomes.
Table 2: Meta-Analysis of Vaccine Platform Efficacy (2020-2025) [7]
| Vaccine Platform | Risk Ratio (RR) | 95% Confidence Interval | Comparative Efficacy |
|---|---|---|---|
| Viral Vector Vaccines | 1.90 | 0.08 – 46.65 | Higher protection, but high variability |
| Virus-Like Particle (VLP) Vaccines | 1.66 | 0.97 – 2.86 | Higher protection |
| Peptide-Based Vaccines | 1.09 | 0.75 – 1.57 | Moderate efficacy |
| Dendritic Cell-Based Vaccines | Not specified | Not specified | Limited benefit |
Beyond efficacy, optimizing transduction protocols can yield significant quantitative improvements. Research in 2025 demonstrated that a sequential treatment with polybrene (PB) and hydroxychloroquine (HCQ) enhanced AAV transduction efficiency in 3D organoid models by approximately 1.3- to 2-fold compared to single-agent treatments, and 1.7- to 2.5-fold compared to virus alone [5] [6]. This enhancement, achieved while maintaining cell viability above 80-90%, provides a clear methodology for overcoming barriers in complex 3D systems.
Enhanced Protocol for AAV Transduction in 3D Organoids
This optimized protocol leverages the synergistic effect of polybrene (PB), which facilitates viral entry by reducing electrostatic repulsion, and hydroxychloroquine (HCQ), which modulates endosomal processing and TLR9-mediated innate immune responses [5] [6]. The sequential administration targets distinct stages of the viral transduction pathway, significantly improving efficiency in structurally complex systems.
Materials and Reagents
Table 3: Research Reagent Solutions for Enhanced AAV Transduction
| Reagent / Material | Function / Application | Working Concentration |
|---|---|---|
| Polybrene (PB) | Cationic polymer that enhances viral entry by neutralizing charge repulsion [5] | 8-10 μg/mL |
| Hydroxychloroquine (HCQ) | Modulates endosomal processing and inhibits TLR9-mediated immune responses [5] | 15-20 μM |
| AAV Vectors (e.g., mCherry) | Gene delivery vehicle; validate titer and purity (empty/full capsid ratio) prior to use [4] | MOI 2×10^4 |
| Liver/Retinal Organoids | 3D model system; culture using established protocols [5] | N/A |
| Cell Viability Assay (CCK-8) | Assess cytotoxicity of treatment conditions [6] | N/A |
| TUNEL Assay Kit | Confirm preserved cellular integrity post-treatment [5] | N/A |
Step-by-Step Procedure
Preparation and Pre-treatment:
- Culture liver or retinal organoids using established protocols and validate at defined maturation stages [5].
- Prepare a working solution of PB at 8 μg/mL for liver organoids or 10 μg/mL for retinal organoids in the appropriate culture medium.
- Pretreat organoids with the PB solution for 4 hours at 37°C, 5% CO₂ [6].
Viral Transduction:
- Dilute the AAV vector (e.g., mCherry reporter) in fresh culture medium to a multiplicity of infection (MOI) of 2 × 10⁴.
- Remove the PB-containing medium from the organoids.
- Add the virus-containing medium to the organoids and incubate for the duration specified for your specific AAV serotype and organoid model.
Post-treatment with HCQ:
- Prepare a working solution of HCQ at 20 μM for liver organoids or 15 μM for retinal organoids in culture medium.
- Following viral transduction, replace the virus-containing medium with the HCQ solution.
- Incubate liver organoids with HCQ for 36 hours and retinal organoids for 48 hours [5].
Analysis and Validation:
- After the HCQ post-treatment, replace with standard culture medium.
- Analyze transduction efficiency via confocal microscopy (e.g., mCherry fluorescence) on day 10 (liver) or day 15 (retinal) post-transduction.
- Quantify the mCherry-positive area or mean fluorescence intensity using image analysis software.
- Confirm minimal cytotoxicity and preserved cellular integrity using a TUNEL assay and cell viability assay (CCK-8), expecting TUNEL-positive cells to remain low (1.1–3.9%) with no significant differences from controls [5] [6].
Diagram 1: AAV Transduction Enhancement Workflow.
Expected Results and Quality Control
Implementation of this sequential protocol should yield a 1.3- to 2-fold increase in transduction efficiency compared to single-agent treatments, and a 1.7- to 2.5-fold increase compared to virus-only controls in both liver and retinal organoid models [5]. Bright-field imaging should confirm no adverse changes in organoid morphology or density. Flow cytometry and quantitative image analysis will validate the significant increase in both the proportion of transduced cells and the intensity of transgene expression (e.g., *p < 0.0001 and *p < 0.01 compared to virus-only groups) [6]. Crucially, cell viability should remain ≥80%, and TUNEL assays should show minimal apoptosis (1.1–3.9%), with no statistically significant differences from untreated controls [5].
Advanced Monitoring and Safety Assessment Protocol
For integrating vectors like lentivirus and gamma-retrovirus, monitoring insertion sites is critical for assessing long-term safety. The MELISSA (ModELing IS for Safety Analysis) framework provides a statistical approach for analyzing integration site (IS) data to quantify insertional mutagenesis risk [3].
Materials and Reagents
- Integration Site (IS) Data: Provided in BED file format, containing clone size estimates (read counts, UMIs, or shear site data) [3].
- Design Matrix: A file containing sample-specific covariates (e.g., condition, replicate, cell type, time post-therapy).
- Genome Annotation File: Standard file (e.g., GTF) for the reference genome.
- MELISSA R Package: The core statistical tool for analysis [3].
Step-by-Step Procedure
Data Input and Preparation:
- Compile IS tables from sequencing data in the required BED format.
- Create a design matrix specifying the experimental conditions and time points for all samples.
Statistical Modeling with MELISSA:
- Perform gene targeting rate analysis using logistic regression to identify genomic regions preferentially targeted by integration events. This model tests whether the observed IS frequency in a given gene deviates significantly from the genome-wide average.
- Perform clone fitness analysis to evaluate whether integration within a specific gene influences clonal expansion over time. This model tests if clones with IS in a given gene show growth rates different from the global baseline.
Downstream Analysis and Interpretation:
- Generate descriptive statistics and clonality indexes.
- Perform gene set enrichment analysis (GSEA) on results to identify biological pathways potentially affected by insertional mutagenesis.
- Use the model outputs—Likelihood Ratio Test (LRT) statistics and regression coefficients—to identify genes with significant targeting or growth effects.
Diagram 2: Integration Site Analysis Workflow.
Expected Results and Quality Control
Application of MELISSA to preclinical or clinical IS data should successfully identify both known and novel genes associated with altered clonal fitness upon vector integration [3]. The framework is sensitive enough to detect early signs of clonal expansion, even in datasets without overtly concerning clonal abundances (e.g., with dominant clones representing only 1-9.5% of the population) [3]. Performance metrics from simulation studies, including Positive Predictive Value (PPV) and detection rates, should be evaluated to ensure statistical power given the sample size and effect size of the experiment.
The clinical translation of viral vector-based therapies necessitates overcoming significant hurdles in manufacturing, safety, and efficient gene delivery to complex physiological models. The protocols detailed herein—a combinatorial chemical treatment to enhance AAV transduction in 3D organoids and a robust statistical framework for assessing insertional mutagenesis risk—provide researchers with actionable strategies to address these imperatives. By adopting these optimized methods, scientists can improve the efficiency and predictive power of preclinical studies, contribute to the development of safer vector designs, and ultimately accelerate the advancement of reliable and accessible gene therapies.
Gene therapy holds immense potential for treating genetic disorders, malignancies, and infectious diseases through the targeted introduction, silencing, or precise editing of therapeutic genes [8]. The clinical success of these advanced therapies is fundamentally constrained by the delivery vehicles, or vectors, used to transport genetic cargo into target cells. While viral vectors have historically dominated therapeutic applications due to their high transduction efficiency, they present significant challenges including robust immunogenicity, insertional mutagenesis risks, and limited cargo capacity [8] [1]. Non-viral nanoparticle vectors have consequently emerged as promising alternatives, offering superior safety profiles, manufacturing scalability, and expanded structural and functional reconfigurability to accommodate various cargo sizes [8].
The development of non-viral gene delivery systems represents a paradigm shift in therapeutic gene transfer, particularly with the advent of CRISPR-based gene editing technologies that require precise, transient delivery of editing components [9]. Nanoparticles, defined as particles with dimensions approximately 10⁻⁹ meters, exhibit distinctive behaviors due to their high surface area-to-mass ratios, enabling enhanced colloidal stability, novel electrical properties, and customizable surface characteristics [9]. These physicochemical properties make nanoparticles particularly suitable for overcoming the biological barriers to gene delivery, including nuclease-mediated degradation, cellular uptake limitations, and intracellular trafficking obstacles [10] [9].
This application note provides a comprehensive technical resource for researchers, scientists, and drug development professionals working in gene therapy. We present structured quantitative comparisons, detailed experimental protocols, and visualization tools to facilitate the implementation of non-viral nanoparticle platforms in research and therapeutic development, with particular emphasis on their core advantages in safety, scalability, and cargo capacity.
Quantitative Advantage Analysis
The comparative advantages of non-viral nanoparticle systems across safety, scalability, and cargo capacity parameters can be quantitatively assessed against viral vector systems. The data presented in the following tables provide a structured framework for objective evaluation during vector selection.
Table 1: Comparative Analysis of Vector Systems Based on Core Advantages
| Evaluation Parameter | Viral Vector Systems | Non-Viral Nanoparticle Systems |
|---|---|---|
| Safety Profile | • Immunogenicity: Moderate to High (e.g., Adenovirus: High; AAV: Low-Moderate) [1] [10]• Genomic Integration: Risk with Lentivirus/Retrovirus, leading to potential insertional mutagenesis [1] [10]• Pre-existing Immunity: Common for AAV and Adenovirus, may limit efficacy [1] | • Immunogenicity: Generally Low [8] [11]• Genomic Integration: Typically non-integrating, significantly reducing mutagenesis risk [8]• Toxicity Concerns: Primarily related to carrier material (e.g., cationic lipid/polymer toxicity) [9] |
| Scalability & Manufacturing | • Production Complexity: High; requires cell culture, purification from viral components [12]• Process Duration: Potentially months-long process with heterogeneous product yields [12]• Cost: High cost of goods (COGs) [1] | • Production Complexity: Low to Moderate; often uses scalable chemical synthesis [8]• Process Scalability: Highly scalable and reproducible manufacturing [8] [13]• Cost: Lower COGs compared to viral vectors [8] |
| Cargo Capacity | • Strict Limitations: AAV: ~4.7 kb [1] [11]; Lentivirus: ~8 kb [10]• Large Gene Challenge: Incompatible with large genetic elements without complex splitting strategies [1] | • Flexible & Large Capacity: Can be engineered to accommodate large DNA, mRNA, or RNP complexes (e.g., >10 kb) [8] [9]• Co-delivery Capability: Can deliver multiple therapeutic agents (e.g., Cas9 protein + gRNA + donor DNA) simultaneously [9] |
| Therapeutic Examples | • AAV: Luxturna, Zolgensma [1]• Lentivirus: CAR-T therapies (Kymriah, Zynteglo) [1] | • LNP: Patisiran (Onpattro) [1]• GalNAc-siRNA: Givosiran (Givlaari), Lumasiran (Oxlumo) [1] |
Table 2: Cargo Capacity and Characteristics of Non-Viral Nanoparticle Systems
| Cargo Type | Typical Size Range | Key Advantages | Ideal Applications |
|---|---|---|---|
| Plasmid DNA (pDNA) | 3 - 20 kbp | Stable, long-term production of template for gene addition [10] | Gene replacement, long-term transgene expression |
| mRNA | 1 - 5 kb | Transient expression, no risk of genomic integration, rapid protein production [8] [11] | Vaccines, transient gene expression, CRISPR-Cas9 editing |
| Ribonucleoprotein (RNP) | ~160 kDa (Cas9) + gRNA | Immediate activity, highest precision, reduced off-target effects, shortest cellular residence [11] | CRISPR-based gene editing (knockout, knock-in) |
| Small RNA (siRNA, miRNA) | 19 - 25 bp (duplex) | Efficient gene silencing, well-established delivery chemistries (e.g., GalNAc) [1] | Gene silencing, target validation |
Table 3: Market and Clinical Adoption Trends (2024-2035 Projections)
| Metric | Viral Gene Delivery | Non-Viral Gene Delivery |
|---|---|---|
| Market Share (2025) | ~66% [13] | ~34% (Including other non-viral) [13] |
| Projected CAGR (2025-2035) | 7.7% [13] | 5.8% (Segment including chemical/physical methods) [13] |
| Key Growth Driver | High efficiency in established therapies (e.g., CAR-T, monogenic diseases) [13] | Demand for safer, scalable platforms for CRISPR, mRNA vaccines, and personalized medicine [8] [13] |
Experimental Protocols
Protocol: Formulation of Lipid Nanoparticles (LNPs) for mRNA Encapsulation
This protocol describes a standardized method for preparing LNPs using microfluidic mixing, suitable for encapsulating mRNA cargo for in vitro and in vivo delivery applications [11] [9].
Research Reagent Solutions:
- Ionizable Cationic Lipid: e.g., DLin-MC3-DMA or SM-102; forms the core structure and enables endosomal escape.
- Helper Phospholipid: e.g., DSPC; enhances bilayer stability and fusogenicity.
- Cholesterol: Modulates membrane fluidity and stability.
- PEGylated Lipid: e.g., DMG-PEG2000; reduces particle aggregation and opsonization, improves stability.
- mRNA Cargo: Purified in vitro transcribed (IVT) mRNA in citrate buffer (pH 4.0).
- Ethanol (absolute)
- Sodium Acetate Buffer (25 mM, pH 4.0)
- 1x Phosphate Buffered Saline (PBS), pH 7.4
Procedure:
- Lipid Stock Preparation: Prepare the lipid mixture in ethanol at a molar ratio of 50:10:38.5:1.5 (Ionizable Lipid:DSPC:Cholesterol:PEG-Lipid) with a total lipid concentration of 12.5 mM. Warm slightly if needed to ensure complete dissolution.
- Aqueous Phase Preparation: Dilute the mRNA cargo in 25 mM sodium acetate buffer (pH 4.0) to a final concentration of 0.1 mg/mL.
- Microfluidic Mixing:
- Use a commercial microfluidic mixer (e.g., NanoAssemblr) or a custom-made setup.
- Set the flow rate ratio (Aqueous:Ethanol) to 3:1.
- Simultaneously pump the aqueous mRNA solution and the ethanolic lipid solution into the mixing chamber at a combined total flow rate of 12 mL/min.
- Collect the resulting LNP suspension in a sterile vial.
- Buffer Exchange and Dialysis:
- Immediately dilute the formed LNPs with an equal volume of 1x PBS (pH 7.4).
- Transfer the solution to a dialysis cassette (e.g., 20 kDa MWCO) and dialyze against a >100x volume of 1x PBS for 4-6 hours at 4°C, with one buffer change.
- Characterization:
- Size and PDI: Measure by Dynamic Light Scattering (DLS). Target size: 70-100 nm; PDI: <0.2.
- Zeta Potential: Measure by Laser Doppler Velocimetry. Expected value: Slightly negative to neutral in PBS.
- Encapsulation Efficiency: Quantify using a Ribogreen assay. Compare fluorescence with and without a detergent (Triton X-100) to distinguish encapsulated vs. free mRNA. Target: >90%.
Protocol: Preparation of CRISPR Ribonucleoprotein (RNP) Complexes
This protocol details the formation of Cas9-gRNA ribonucleoprotein (RNP) complexes, a preferred cargo for precise genome editing due to rapid activity and minimal off-target effects [11].
Research Reagent Solutions:
- Recombinant Cas9 Nuclease: High-purity, endotoxin-free, resuspended in storage buffer.
- Target-specific sgRNA: Chemically synthesized or in vitro transcribed, HPLC-purified.
- Nuclease-Free Duplex Buffer: (e.g., 30 mM HEPES, pH 7.5, 100 mM Potassium Acetate).
- Nuclease-Free Water.
Procedure:
- sgRNA Preparation: Centrifuge the sgRNA tube briefly and resuspend in nuclease-free duplex buffer to a stock concentration of 160 µM. Store on ice.
- Complex Formation:
- Prepare the RNP complex in a nuclease-free microcentrifuge tube.
- For a 10 µL reaction, use a 1:1.2 molar ratio of Cas9 to sgRNA. A typical setup is:
- Cas9 (40 µM stock): 5 µL (200 pmol)
- sgRNA (160 µM stock): 1.5 µL (240 pmol)
- Nuclease-Free Duplex Buffer: 3.5 µL
- Gently pipette to mix. Do not vortex.
- Incubation: Incubate the mixture at room temperature (25°C) for 10-20 minutes to allow for complete RNP complex formation.
- Quality Control (Optional):
- Analyze complex formation using a native agarose gel (1-2%) or a gel shift assay. The RNP complex should show a mobility shift compared to free Cas9 protein.
- Immediate Use: The formed RNP complexes should be used immediately for transfection or can be stored on ice for a short period (<1 hour) before encapsulation into nanoparticles.
Protocol: Characterization of Nanoparticle Cytotoxicity and Immunogenicity
This protocol outlines standardized in vitro assays to evaluate the safety profile of formulated nanoparticles, a critical step in preclinical development [12] [9].
Research Reagent Solutions:
- Cell Line: Relevant human cell line (e.g., HEK293, HepG2, primary fibroblasts).
- Cell Culture Media: Appropriate complete media for the selected cell line.
- Test Nanoparticles: Formulated nanoparticles in PBS, sterile-filtered.
- MTT Reagent: (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) at 5 mg/mL in PBS.
- Dimethyl Sulfoxide (DMSO)
- ELISA Kit for Human TNF-α or IL-6
- Flow Cytometry Staining Buffer (PBS + 1% BSA)
- Antibodies: Anti-human CD80, CD86, and MHC Class II, with appropriate isotype controls.
Procedure: Part A: Cell Viability Assay (MTT)
- Seed cells in a 96-well plate at a density of 1x10⁴ cells/well and incubate for 24 hours.
- Treat cells with a serial dilution of nanoparticles (e.g., 0, 10, 25, 50, 100 µg/mL total lipid) in triplicate. Include a vehicle control (PBS) and a positive control for death (e.g., 1% Triton X-100).
- Incubate for 24-48 hours.
- Add 20 µL of MTT reagent to each well and incubate for 3-4 hours at 37°C.
- Carefully remove the media and solubilize the formed formazan crystals with 150 µL of DMSO.
- Measure the absorbance at 570 nm using a plate reader. Calculate cell viability as a percentage of the vehicle control.
Part B: Innate Immune Response Profiling
- For cytokine release, seed and treat cells as in Part A, using a relevant immune cell model like peripheral blood mononuclear cells (PBMCs) or THP-1-derived macrophages.
- After 24 hours of nanoparticle exposure, collect the cell culture supernatant by centrifugation.
- Analyze the supernatant for pro-inflammatory cytokines (e.g., TNF-α, IL-6) using a commercial ELISA kit, following the manufacturer's instructions.
- For surface activation marker analysis, harvest the cells after treatment, wash with staining buffer, and stain with fluorescently-labeled antibodies against CD80, CD86, and MHC Class II for 30 minutes on ice in the dark.
- Wash cells and resuspend in staining buffer for analysis by flow cytometry. Compare the median fluorescence intensity (MFI) of treated cells to untreated controls to assess immune activation.
Visualizing Workflows and Mechanisms
LNP Formulation and Intracellular Delivery
Diagram Title: LNP Formulation and Delivery Pathway
CRISPR-Cas9 RNP Delivery and Editing
Diagram Title: RNP Delivery and Gene Editing Mechanism
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for Non-Viral Vector Research
| Reagent / Material | Function / Application | Key Characteristics |
|---|---|---|
| Ionizable Cationic Lipids (e.g., DLin-MC3-DMA, SM-102) | Core component of LNPs; self-assembles with nucleic acids, enables endosomal escape via proton sponge effect [11] [9]. | pKa ~6.5, biodegradable ester linkages, high fusogenicity. |
| Cationic Polymers (e.g., Polyethylenimine - PEI) | Condenses nucleic acids via electrostatic interaction; common non-viral vector for in vitro transfection [8] [10]. | High positive charge density, proton sponge effect; can be cytotoxic. |
| PEGylated Lipids (e.g., DMG-PEG2000) | Provides a hydrophilic corona on nanoparticles; reduces aggregation, improves stability, and prolongs circulation half-life in vivo [9]. | PEG chain length (e.g., 2000 Da) critical for steric stabilization. |
| Recombinant Cas9 Nuclease | Engineered CRISPR-associated protein; functions as molecular scissors for targeted DNA cleavage in RNP complexes [11] [9]. | High purity, endotoxin-free, nuclear localization signals (NLS), various fidelity versions available. |
| Synthetic sgRNA | Single guide RNA; directs Cas9 to specific genomic loci via complementary base pairing [11]. | Chemically modified (e.g., 2'-O-methyl) for enhanced nuclease resistance and reduced immunogenicity. |
| Microfluidic Mixer (e.g., NanoAssemblr) | Enables rapid, reproducible, and scalable mixing of aqueous and organic phases for homogeneous nanoparticle formation [9]. | Precise control over particle size and PDI; compatible with GMP production. |
Non-viral nanoparticle vectors represent a transformative platform in gene therapy, decisively addressing the critical limitations of viral vectors through enhanced safety, scalable manufacturing, and superior cargo flexibility. The structured data, protocols, and visualizations provided in this application note equip researchers with the foundational tools to leverage these advantages. As the field progresses, the convergence of novel biomaterials, advanced targeting strategies, and deep biological insight will undoubtedly expand the therapeutic reach of non-viral gene delivery, enabling more effective and accessible treatments for a broad spectrum of human diseases.
Gene therapy represents a transformative approach for treating genetic disorders, malignancies, and infectious diseases through the targeted introduction, silencing, or precise editing of therapeutic genes [8]. The success of these therapies is fundamentally dependent on delivery vectors that can safely and efficiently transport genetic payloads to target cells. Non-viral nanoparticles have emerged as promising alternatives to viral vectors, offering superior safety profiles, scalability for manufacturing, and structural reconfigurability to accommodate various cargo sizes [8] [14]. This application note provides a comprehensive landscape overview of the primary classes of non-viral nanoparticle vectors, detailing their compositions, mechanisms, applications, and experimental protocols relevant to gene delivery research and therapeutic development.
Comparative Analysis of Non-Viral Nanoparticle Classes
Table 1: Key Characteristics of Major Non-Viral Nanoparticle Classes
| Nanoparticle Class | Key Components | Mechanism of Action | Advantages | Limitations | Therapeutic Applications |
|---|---|---|---|---|---|
| Lipid Nanoparticles (LNPs) | Ionizable lipids, phospholipids, cholesterol, PEG-lipids [15] | pH-dependent charge reversal; endosomal escape via disruption [15] | High encapsulation efficiency; proven clinical success; biocompatible [16] | Predominant liver tropism; complex optimization [15] | siRNA drugs (Onpattro), mRNA vaccines, CRISPR delivery [15] [1] |
| Polymeric Nanoparticles | PEI, PAMAM, PLGA, chitosan [17] | DNA condensation via electrostatic interaction; "proton sponge" endosomal escape [17] | Structural diversity; facile functionalization; sustained release [17] | Potential cytotoxicity; aggregation issues [17] | Cardiovascular disease treatment, cancer therapy [17] |
| Liposomes | Phospholipids, cholesterol [16] | Lipid bilayer encapsulation; membrane fusion [16] | Biocompatibility; hydrophilic/hydrophobic payload capacity [16] | Low nucleic acid encapsulation efficiency [16] | Drug delivery, limited gene therapy applications [16] |
| Inorganic Nanoparticles | Gold, iron oxide, silica [18] | Variable based on material (e.g., magnetic targeting, thermal responsiveness) [18] | Tunable physicochemical properties; diagnostic and therapeutic multifunctionality [18] | Potential long-term toxicity concerns; biodegradability challenges [18] | Imaging, diagnostics, hyperthermia-based therapies [18] |
Composition and Structure-Function Relationships
Lipid Nanoparticles (LNPs)
LNPs represent the most clinically advanced non-viral gene delivery platform, typically consisting of four key lipid components [15]:
Ionizable Lipids: Critical for nucleic acid encapsulation and endosomal escape. These lipids possess a pKa typically between 6.2-6.9, enabling positive charge acquisition in acidic environments for RNA complexation while maintaining neutrality at physiological pH to reduce toxicity [15]. Structurally, they consist of an amine head group connected to hydrophobic tails via biodegradable linkers (ester, ether, or amide bonds) [15].
Phospholipids (e.g., DSPC, DOPE): Stabilize the LNP bilayer structure and contribute to endosomal escape. DSPC is often preferred for siRNA delivery, while DOPE demonstrates superior performance for mRNA encapsulation [15].
Cholesterol: Enhances membrane integrity and stability of LNPs during circulation [15].
PEG-lipids: Constitute approximately 1.5 mol% of formulation but critically impact stability through steric hindrance, reducing aggregation and protein adsorption [15].
Polymeric Nanoparticles
Cationic polymers facilitate nucleic acid complexation through electrostatic interactions between polymer amine groups and nucleotide phosphate groups [17]. Key polymeric systems include:
Polyethylenimine (PEI): Considered the "gold standard" polymeric vector, available in linear and branched architectures. PEI facilitates endosomal escape via the "proton sponge" effect but can exhibit significant cytotoxicity [17].
Polyamidoamine (PAMAM): Dendrimeric structure offering precise molecular architecture for functionalization. Surface modification with targeting ligands (e.g., antibodies) enhances specificity [17].
Poly(lactic-co-glycolic acid) (PLGA): Biodegradable polyester with excellent biocompatibility, enabling sustained release applications [17].
Liposomes vs. Lipid Nanoparticles
While both are lipid-based systems, fundamental differences exist. Liposomes feature simple phospholipid bilayers with neutral charge, resulting in poor nucleic acid encapsulation efficiency [16]. LNPs incorporate ionizable cationic lipids that enhance RNA interaction and encapsulation while maintaining physiological compatibility [16].
Targeting Strategies and Surface Modification
Table 2: Nanoparticle Surface Modification Strategies for Enhanced Targeting
| Modification Approach | Specific Ligands/Strategies | Mechanism | Target Sites | Impact on Delivery Efficiency |
|---|---|---|---|---|
| Active Targeting Ligands | Antibodies, peptides, aptamers, small molecules (e.g., folate, galactose) [15] [17] | Receptor-ligand binding promoting cellular uptake | Cell-specific receptors (e.g., asialoglycoprotein receptor for GalNAc) [1] | Enhanced cellular specificity and uptake; reduced off-target effects [15] |
| Stealth Coatings | PEGylation, chitosan [18] | Steric hindrance reducing protein adsorption and immune recognition | Systemic circulation | Prolonged circulation half-life; reduced clearance by mononuclear phagocyte system [18] |
| Formulation Optimization | Component ratio adjustment; novel lipid design [15] | Altering physicochemical properties (size, charge, pKa) | Specific tissues/organs beyond liver | Modulated biodistribution; enhanced endosomal escape [15] |
| Stimuli-Responsive Elements | pH-sensitive lipids, enzyme-cleavable linkers [19] | Payload release triggered by specific biological stimuli | Disease microenvironments (e.g., acidic tumors) | Controlled spatiotemporal release; enhanced therapeutic precision [19] |
Overcoming Biological Barriers
Effective gene delivery requires nanoparticles to overcome multiple biological barriers [16]:
Systemic Barriers: Serum nucleases rapidly degrade naked nucleic acids, while the mononuclear phagocyte system clears circulating nanoparticles. PEGylation creates a hydrophilic "protective barrier" that reduces protein adsorption and extends circulation time [18].
Cellular Barriers: The negatively charged cell membrane repels nucleic acids. Nanoparticles with positive surface charge (zeta potential) facilitate cellular uptake through electrostatic interactions, though excessive positivity increases toxicity [16]. Optimal nanoparticle size (60-100 nm) promotes receptor-mediated endocytosis while avoiding renal clearance (<10 nm) or immune activation (>300 nm) [16].
Intracellular Barriers: Following endocytosis, nanoparticles must escape endosomal compartments before enzymatic degradation. Ionizable lipids and protonable polymers facilitate endosomal membrane disruption through charge reversal in acidic environments [15] [17].
Experimental Protocols
LNP Formulation via Microfluidic Mixing
Principle: Rapid mixing of lipid and aqueous phases induces nanoprecipitation, forming monodisperse LNPs [15].
LNP Formulation Workflow
Materials:
- Ionizable lipid (e.g., DLin-MC3-DMA, SM-102)
- Helper lipids: DSPC, cholesterol
- PEG-lipid (e.g., DMG-PEG 2000)
- Anhydrous ethanol
- mRNA in 10 mM citrate buffer (pH 4.0)
- Microfluidic device (NanoAssemblr, Precision NanoSystems)
- Dialysis membranes (MWCO 100 kDa)
- Phosphate-buffered saline (PBS, pH 7.4)
Procedure:
- Lipid Solution Preparation: Dissolve ionizable lipid, DSPC, cholesterol, and PEG-lipid in ethanol at molar ratio 50:10:38.5:1.5 to achieve total lipid concentration of 10-20 mM [15].
- Aqueous Phase Preparation: Dilute mRNA in citrate buffer to concentration of 0.1-0.2 mg/mL.
- Microfluidic Mixing: Set total flow rate (TFR) to 12 mL/min with aqueous-to-organic flow rate ratio (FRR) of 3:1.
- Immediate Dialysis: Dialyze formed LNPs against PBS (pH 7.4) for 4 hours at 4°C to remove ethanol.
- Characterization: Measure particle size (target: 60-100 nm), polydispersity index (PDI <0.2), and encapsulation efficiency (>90%) [16].
Polymer-DNA Polyplex Formation
Principle: Electrostatic complexation between cationic polymer and anionic DNA forms compact nanoparticles [17].
Polyplex Formation Protocol
Materials:
- Branched PEI (25 kDa, Sigma-Aldrich)
- Plasmid DNA (pDNA) encoding therapeutic gene
- HEPES-buffered glucose (HBG, pH 7.4)
- Agarose gel electrophoresis equipment
Procedure:
- Polymer Solution: Prepare PEI at 1 mg/mL in HBG buffer, filter sterilize (0.22 μm).
- DNA Dilution: Dilute pDNA to 0.1 mg/mL in HBG buffer.
- Polyplex Formation: Add polymer solution to DNA solution at specified N/P ratios (typically 5-10:1, amine to phosphate). Vortex immediately for 30 seconds.
- Incubation: Allow polyplex formation to complete by incubating 30 minutes at room temperature.
- Characterization:
- Size and zeta potential: Dynamic light scattering
- Complexation efficiency: Agarose gel electrophoresis retardation assay
- Morphology: Transmission electron microscopy
In Vitro Transfection Efficiency Assessment
Principle: Quantitative measurement of gene expression following nanoparticle-mediated delivery [19].
Materials:
- HEK293 or HeLa cells
- Complete growth medium (DMEM + 10% FBS)
- Reporter plasmid (e.g., eGFP, luciferase)
- Formulated nanoparticles
- Flow cytometer or luminometer
- MTT assay reagents for cytotoxicity
Procedure:
- Cell Seeding: Plate cells in 24-well plates at 50,000 cells/well, incubate 24 hours to reach 70-80% confluency.
- Nanoparticle Treatment: Replace medium with fresh complete medium containing nanoparticles at serial dilutions. Include positive (commercial transfection reagent) and negative (untreated cells) controls.
- Incubation: Incubate cells 24-48 hours at 37°C, 5% CO₂.
- Efficiency Analysis:
- For eGFP expression: Analyze using flow cytometry, reporting percentage of fluorescent cells and mean fluorescence intensity.
- For luciferase expression: Lyse cells, measure luminescence, normalize to protein content.
- Cytotoxicity Assessment: Perform MTT assay parallel to transfection to determine cell viability.
Table 3: Research Reagent Solutions for Non-Viral Gene Delivery
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Ionizable Lipids | DLin-MC3-DMA, SM-102, ALC-0315 [15] | Core LNP component for nucleic acid encapsulation and endosomal escape | pKa optimization (6.2-6.9); biodegradability via ester bonds [15] |
| Cationic Polymers | PEI (branched, 25kDa), PAMAM dendrimers [17] | DNA condensation; proton sponge endosomal escape | Cytotoxicity concerns; requires structural modification [17] |
| PEG-Lipids | DMG-PEG2000, DSG-PEG2000 [15] | LNP stability; reduced protein adsorption; circulation half-life extension | PEG content optimization (typically 1.5 mol%); potential anti-PEG immunity [15] |
| Helper Lipids | DSPC, DOPE, cholesterol [15] | LNP structural integrity; membrane fluidity modulation | DOPE preferred for mRNA; DSPC for siRNA [15] |
| Targeting Ligands | GalNAc, RGD peptides, transferrin, folate [15] [17] [18] | Cell-specific targeting through receptor recognition | Conjugation chemistry; ligand density optimization [18] |
| Characterization Tools | DLS, zeta potential analyzer, TEM [16] | Nanoparticle physicochemical property assessment | Size (60-100 nm optimal); PDI (<0.2); zeta potential (moderate positive) [16] |
The landscape of non-viral nanoparticles for gene delivery encompasses diverse platforms with complementary strengths and applications. LNPs currently lead clinical translation with proven success in siRNA and mRNA delivery, while polymeric nanoparticles offer extensive functionalization flexibility. Liposomes provide established biocompatibility, and inorganic nanoparticles enable unique theranostic applications. Critical to advancing these platforms is the rational design of nanoparticle composition and surface properties to overcome biological barriers and achieve targeted delivery. The experimental protocols outlined provide foundational methodologies for researchers developing next-generation non-viral gene delivery systems. As these technologies continue to evolve, they hold immense potential to expand the therapeutic reach of gene-based medicines beyond current limitations.
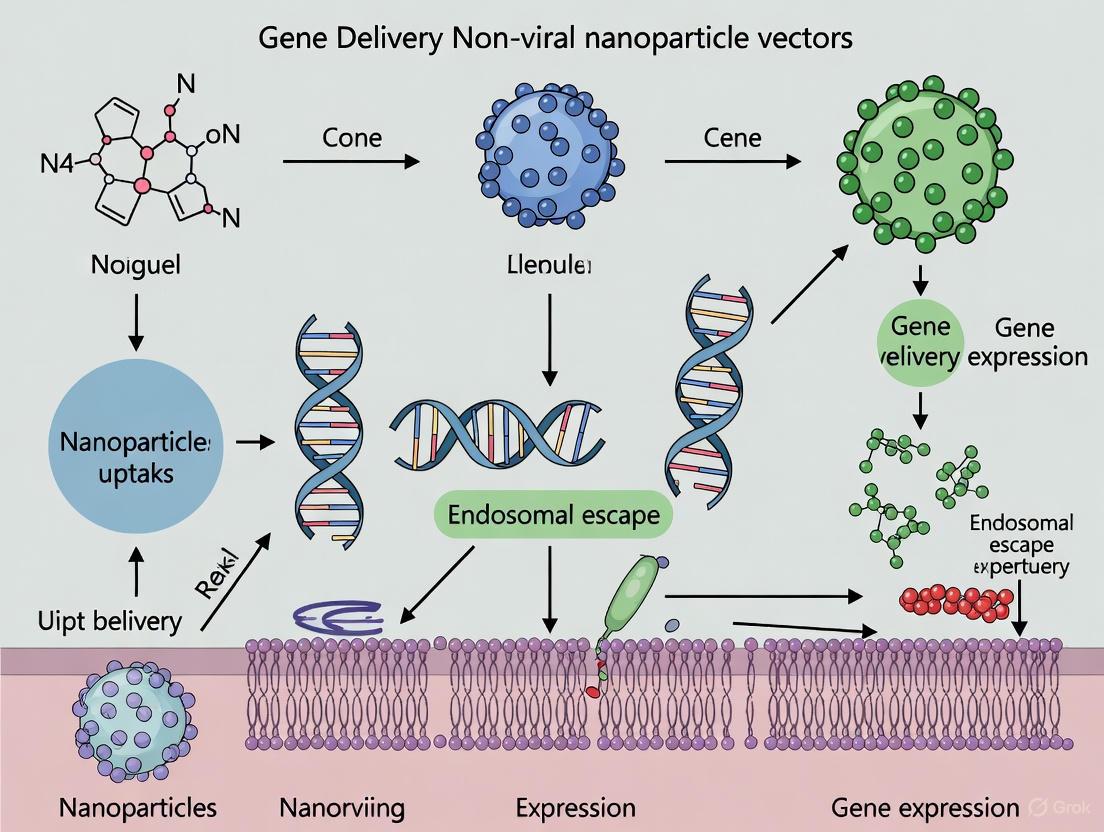
Mechanisms of Cellular Uptake and Intracellular Trafficking
The efficacy of non-viral nanoparticle vectors in gene delivery is fundamentally governed by their journey from initial cellular contact to final intracellular destination. Understanding the mechanisms of cellular uptake and subsequent intracellular trafficking is paramount for the rational design of vectors that can overcome biological barriers and achieve therapeutic levels of gene expression. This document provides detailed application notes and protocols to study these critical processes, framed within a thesis investigating poly(beta-amino ester) (PBAE) polymers as model non-viral vectors for glioblastoma gene therapy [20]. The following sections outline the primary internalization pathways, provide a quantitative framework for analyzing plasmid mass transfer, and detail essential reagents and protocols for experimental investigation.
Major Uptake Pathways for Non-Viral Nanoparticles
Non-viral gene complexes, or polyplexes, are internalized via a variety of endocytic and non-endocytic pathways. The dominant route depends on a complex interplay of nanoparticle physicochemical properties and the target cell type [21]. Table 1 summarizes the characteristics of the key endocytic pathways.
Table 1: Key Endocytic Pathways for Non-Viral Nanoparticle Vectors
| Uptake Pathway | Key Machinery/Features | Typical Cargo Size | Intracellular Fate | Common Inhibitors |
|---|---|---|---|---|
| Clathrin-Mediated Endocytosis (CME) | Clathrin coat, dynamin [21] | ~120 nm [21] | Early endosome → late endosome → lysosome [21] | Chlorpromazine, Pitstop 2 [21] |
| Caveolae-Mediated Endocytosis (CvME) | Caveolin-1, cholesterol-rich domains, dynamin [21] | ~60 nm [21] | Caveosome → Endoplasmic Reticulum/Golgi [21] | Methyl-β-cyclodextrin, Genistein [21] |
| Macropinocytosis | Actin-driven membrane ruffling, growth factor receptors [21] | >0.5 μm [21] | Macropinosome → lysosome [21] | Amiloride, EIPA [21] |
| Phagocytosis | Professional phagocytes (e.g., macrophages) [21] | >0.5 μm [21] | Phagosome → lysosome [21] | Cytochalasin D [21] |
The following diagram illustrates the major uptake pathways and the subsequent intracellular trafficking of non-viral nanoparticles, highlighting key compartments and fate decisions.
Quantitative Analysis of Cellular and Nuclear Uptake
A critical bottleneck in non-viral gene delivery is the inefficient transport of genetic material from the cell surface into the nucleus. A quantitative, multi-compartment model using flow cytometry and qPCR can be employed to determine the rate constants for each step, thereby identifying the primary barriers for a given vector system [20].
Protocol: Quantifying Uptake Rates via Flow Cytometry and qPCR
This protocol describes a method to track the number of plasmids through different cellular compartments over time to calculate key rate constants.
Materials
- Cells: Human primary glioblastoma cells [20].
- Vector: Poly(beta-amino ester) polymer (e.g., PBAE 447) [20].
- Plasmid DNA: eGFP-N1 plasmid, conjugated with Cy3 fluorescent dye using a Label IT Tracker Kit [20].
- Buffers: Lysis buffer for nucleus isolation, DNase-free PBS.
- Equipment: Flow cytometer, quantitative PCR machine, NanoDrop spectrophotometer.
Procedure
- Polyplex Formation: Formulate polyplexes at an optimal N/P ratio (e.g., 8.0 for PEI/DNA systems [22]) using Cy3-labeled plasmid DNA. Incubate for a standardized time (e.g., 15-30 minutes) at room temperature to allow for complex formation.
- Cell Transfection: Seed cells at a high density (optimized for the cell type). Add polyplexes to the cells and incubate for defined time points (e.g., 0.5, 1, 2, 4, 8, 24 hours).
- Compartmental Separation:
- Total Cellular Uptake: At each time point, trypsinize and wash cells. Analyze by flow cytometry to measure total Cy3 fluorescence, representing plasmids associated with the cell (Pcell).
- Nuclear Envelope Association & Nuclear Uptake: Lyse the cell membrane using a mild detergent buffer, leaving nuclei intact. Isolate nuclei by centrifugation. Resuspend the nuclear pellet and analyze an aliquot by flow cytometry. The Cy3 signal from the intact nuclei represents plasmids associated with the nuclear envelope (Pne) and those internalized (Pni).
- DNA Quantification via qPCR: Treat the remaining nuclear sample with DNase to degrade any externally bound, non-internalized plasmid DNA. Subsequently, lyse the nuclei and isolate the protected internalized plasmid DNA. Quantify the absolute number of plasmids using qPCR with standard curves of known plasmid quantities. This value is Pni.
- Data Calculation:
- The number of plasmids in the cytoplasm (Pcyto) is calculated as: Pcell - Pne.
- The number of nuclear-associated plasmids (Pne) is calculated from the flow cytometry data of the pre-DNase nuclei sample, calibrated against the qPCR data.
Data Analysis and Modeling
The data is fitted to a four-compartment, first-order mass-action model to determine the rate constants [20]:
- Cellular Uptake Rate Constant (kcell): Conversion from extracellular to cytoplasmic plasmid.
- Nuclear Envelope Association Rate Constant (kne): Conversion from cytoplasmic to nuclear envelope-associated plasmid.
- Nuclear Internalization Rate Constant (kni): Conversion from nuclear envelope-associated to nuclear internalized plasmid.
Table 2 provides example quantitative data derived from applying this model to PBAE-based polyplexes in glioblastoma cells.
Table 2: Quantitative Uptake Metrics for PBAE/DNA Polyplexes in Glioblastoma Cells
| Parameter | Description | Quantitative Value | Interpretation |
|---|---|---|---|
| kcell | Cellular Uptake Rate Constant | 7.5 × 10-4 hr-1 [20] | Rate-limiting step for the system. |
| % of Added Dose Internalized | Total Cellular Uptake Efficiency | 0.1% [20] | Very low fraction of dose enters the cell. |
| % of Internalized DNA in Nucleus | Nuclear Delivery Efficiency | 12% [20] | Once inside the cell, nuclear delivery is relatively efficient. |
| kni | Nuclear Internalization Rate Constant | 1.1 hr-1 [20] | Faster than cellular uptake. |
| Plasmid Degradation | Fast-phase rate constant | 0.62 hr-1 [20] | Indicates significant intracellular degradation. |
The Scientist's Toolkit: Essential Research Reagents
Table 3 catalogs key reagents and tools essential for investigating the cellular uptake and trafficking of non-viral gene delivery vectors.
Table 3: Key Research Reagent Solutions for Uptake and Trafficking Studies
| Reagent / Tool | Function / Application | Example Use |
|---|---|---|
| Chemical Inhibitors | To selectively block specific endocytic pathways and determine their contribution to uptake. | Chlorpromazine (CME), Methyl-β-cyclodextrin (CvME), Amiloride (Macropinocytosis) [21]. |
| Fluorescent Plasmid Labels (e.g., Cy3) | To tag genetic cargo for visualization and quantification via fluorescence microscopy and flow cytometry. | Conjugation to plasmid DNA for tracking cellular and nuclear uptake over time [20]. |
| Gal8-mRuby Reporter System | A live-cell biosensor that fluoresces upon endosomal disruption, directly reporting endosomal escape efficiency. | Screening hundreds of nanoparticle formulations for their ability to escape the endosome and release cargo into the cytosol [23]. |
| Lanthanide-Doped Nanoparticles | Enables quantitative, multiplexed tracking of different nanoparticle formulations simultaneously in a single system. | Comparing biodistribution and tumor delivery of up to 4 different targeted nanoparticles in a single mouse via ICP-MS [24]. |
| 3D Cell Models (Spheroids/Organoids) | Provides a more physiologically relevant model with cell-cell interactions and barriers to penetration, bridging the gap between 2D culture and in vivo. | Studying nanoparticle penetration depth and distribution in a tissue-like context using high-resolution fluorescence microscopy [25]. |
| Metal Chelator-Lipid Conjugates | Allows positron emission tomography (PET) tracking of lipid nanoparticles in live animals and non-human primates. | Visualizing whole-body trafficking of mRNA LNP vaccines, confirming rapid drainage to lymph nodes after intramuscular injection [26]. |
A meticulous, quantitative understanding of the cellular uptake and intracellular trafficking pathways of non-viral nanoparticles is indispensable for advancing gene delivery systems. The application of the protocols and tools detailed herein—from pathway-specific inhibition studies to sophisticated quantitative modeling of plasmid trafficking—enables researchers to identify the specific rate-limiting steps for their vector system. Integrating these insights with advanced models, such as 3D spheroids and multiplexed in vivo tracking, provides a powerful framework for the rational design of next-generation non-viral vectors with enhanced gene delivery efficacy.
Engineering Delivery Platforms: From Lipid Nanoparticles to Inorganic Systems
Lipid nanoparticles (LNPs) have emerged as the non-viral vector of choice for the delivery of a wide spectrum of nucleic acid therapeutics, fundamentally advancing the fields of gene silencing and gene editing. [27] [28] Their journey from a delivery vehicle for small interfering RNA (siRNA) to a sophisticated platform for Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-based genome editing marks a significant milestone in nanomedicine. LNPs are spherical vesicles, typically 50-120 nm in diameter, composed of a precise mixture of ionizable lipids, phospholipids, cholesterol, and PEGylated lipids. [27] [29] This unique composition enables them to efficiently encapsulate and protect nucleic acid cargo, facilitate cellular uptake, and promote endosomal escape for functional delivery into the cytoplasm. [27] [28] The clinical validation of LNP technology was catalyzed by the approval of Onpattro (patisiran) in 2018, the first siRNA therapeutic for hereditary transthyretin-mediated amyloidosis, and its prominence was further solidified by the global deployment of LNP-formulated mRNA vaccines during the COVID-19 pandemic. [30] [31] [32] This application note details the evolution, current applications, and detailed protocols for utilizing LNPs in siRNA and CRISPR delivery, providing a practical resource for researchers and drug development professionals.
LNP Fundamentals: Composition and Mechanism of Action
The functional properties of LNPs are dictated by their core components, each playing a critical role in structure, stability, and delivery efficiency.
- Ionizable Lipids: The most critical component, these lipids are positively charged at low pH (enabling efficient RNA encapsulation during formulation) but neutral at physiological pH (reducing toxicity). They are also instrumental in mediating endosomal escape through a charge-based mechanism. [27] [29]
- Structural Lipids (e.g., DSPC): These phospholipids contribute to the structural integrity of the LNP bilayer, mimicking natural cell membranes and enhancing fusion with cellular membranes. [29]
- Cholesterol: A stability-enhancing component that fills gaps between lipid molecules, increases membrane rigidity, and improves circulation time. [29]
- PEGylated Lipids: These lipids control particle size and prevent aggregation during storage and in circulation by providing a hydrophilic exterior. They also reduce nonspecific interactions and can influence pharmacokinetics. [27] [29]
The mechanism of LNP-mediated delivery follows a multi-step process: First, the LNP protects its nucleic acid payload from degradation in the bloodstream. Following cellular uptake via endocytosis, the acidic environment of the endosome protonates the ionizable lipids, disrupting the endosomal membrane and releasing the cargo into the cytoplasm, where it can execute its function. [27] [28]
→ Diagram: LNP-Mediated Nucleic Acid Delivery Pathway
LNP Delivery of siRNA Therapeutics
Mechanism and Clinical Success
siRNAs are short double-stranded RNA molecules that harness the RNA interference (RNAi) pathway to selectively silence gene expression. [31] The siRNA is loaded into the RNA-induced silencing complex (RISC), where the guide strand directs RISC to complementary messenger RNA (mRNA) sequences. This leads to the cleavage and degradation of the target mRNA, preventing protein translation. [31] [32] LNPs overcome the major hurdles of siRNA delivery, including enzymatic degradation, renal clearance, and inefficient cellular uptake. [31] The success of this approach is demonstrated by several FDA-approved drugs, with more in clinical trials.
Table 1: FDA-Approved LNP-delivered siRNA Therapeutics
| Therapeutic (Brand Name) | Target / Indication | Key Clinical Trial & Outcome | Approval Year |
|---|---|---|---|
| Patisiran (Onpattro) [30] [32] | Transthyretin (TTR) / hATTR Amyloidosis | Phase III (APOLLO): Improved neuropathy scores [30] | 2018 [31] [32] |
| Givosiran (Givlaari) [30] [32] | Aminolevulinic Acid Synthase 1 / Acute Hepatic Porphyria | Phase III (ENVISION): Reduced porphyria attacks [30] | 2019 [32] |
| Inclisiran (Leqvio) [30] | PCSK9 / Hypercholesterolemia | Phase III (ORION): Sustained LDL-C reduction [30] | 2021 [30] |
Protocol: Formulating siRNA-LNPs for Antiviral Applications
Preclinical studies have demonstrated the efficacy of LNP-delivered siRNA against various viruses, including SARS-CoV-2. [31] The following protocol is adapted from these studies.
Materials:
- siRNA: Chemically modified (e.g., 2'-F, 2'-O-Me) for stability. [31]
- Lipids: Ionizable lipid (e.g., ALC-0315), DSPC, Cholesterol, PEG-lipid (e.g., DMG-PEG2000). [27] [29]
- Buffers: 10 mM Citrate buffer (pH 4.0), 1x PBS (pH 7.4).
- Equipment: Microfluidic mixer (e.g., NanoAssemblr), PD-10 desalting columns, Dynamic Light Scattering (DLS) instrument.
Procedure:
- Lipid Stock Preparation: Prepare individual ethanolic stocks of the lipids. Combine at a molar ratio of 50:10:38.5:1.5 (Ionizable Lipid:DSPC:Cholesterol:PEG-lipid). [31] [28]
- Aqueous Phase Preparation: Dilute the siRNA in citrate buffer (pH 4.0) to a final concentration of 0.2 mg/mL.
- Nanoparticle Formation: Using a microfluidic mixer, rapidly mix the ethanolic lipid solution with the aqueous siRNA solution at a fixed flow rate ratio (typically 3:1 aqueous-to-ethanol) to form LNPs. [28]
- Buffer Exchange & Dialysis: Dialyze the formed LNP suspension against a large volume of 1x PBS (pH 7.4) for at least 4 hours at 4°C to remove ethanol and adjust the pH.
- Characterization:
- Particle Size & PDI: Measure by DLS. Target size: 80-100 nm.
- Encapsulation Efficiency: Quantify using a Ribogreen assay. >90% encapsulation is desirable.
- siRNA Integrity: Verify by gel electrophoresis.
Advancing to CRISPR-Cas Genome Editing with LNP Delivery
The Shift from siRNA to CRISPR Payloads
The modularity of LNPs allows them to be adapted for larger and more complex payloads, most notably for CRISPR-Cas genome editing. [27] CRISPR systems can be delivered in multiple formats, each with distinct advantages. While viral vectors are limited by immunogenicity, payload size, and persistent expression, LNP delivery offers a transient, scalable, and less immunogenic alternative. [27] [33]
Table 2: Comparison of CRISPR Formats for LNP Delivery
| CRISPR Format | Components Delivered | Advantages | Challenges |
|---|---|---|---|
| plasmid DNA (pDNA) [29] | DNA encoding Cas9 and gRNA | Simpler formulation. | Low efficiency; requires nuclear entry; prolonged expression increases off-target risk. [27] |
| mRNA + gRNA [29] | mRNA encoding Cas9 protein + guide RNA | Transient expression; higher efficiency than pDNA. | mRNA immunogenicity; co-encapsulation of two RNA species is complex. [27] |
| Ribonucleoprotein (RNP) [33] [29] | Pre-complexed Cas9 protein and gRNA | Highest safety profile; rapid activity; minimal off-target effects. | Formulation challenges due to protein sensitivity and low negative charge. [33] |
A landmark 2025 study published in Nature Biotechnology demonstrated the power of LNP-delivered RNPs. Researchers engineered a thermostable Cas9 (iGeoCas9) that could withstand LNP formulation stresses. Using tissue-selective LNP formulations, they achieved 19% editing efficiency of the disease-causing SFTPC gene in mouse lung tissue and 31% editing of PCSK9 in the liver after a single intravenous injection, showcasing the therapeutic potential of this approach. [33]
Protocol: Assembling CRISPR RNP-LNPs for In Vivo Editing
This protocol is based on the successful methodology for delivering iGeoCas9 RNPs. [33]
Materials:
- CRISPR RNP: Purified iGeoCas9 (or SpyCas9) protein complexed with synthetic sgRNA at a molar ratio of 1:1.2.
- Specialized Lipids: Include a pH-sensitive PEGylated lipid and a biodegradable cationic lipid for enhanced lung targeting. [33]
- Other reagents: Nuclease-free water, Dulbecco's PBS (DPBS).
Procedure:
- RNP Complex Formation: Incubate the Cas9 protein with a 1.2-fold molar excess of sgRNA in nuclease-free buffer for 10 minutes at room temperature to form the RNP complex.
- Lipid Mixture Preparation: Prepare an ethanolic solution of lipids. For lung-tropic delivery, a formulation incorporating ~10 mol% of a biodegradable cationic lipid (e.g., a SORT molecule) is effective. [33] [29]
- Aqueous Phase Preparation: Dilute the pre-formed RNP complex in citrate buffer (pH 4.0).
- LNP Formulation: Use a microfluidic device to mix the ethanolic lipids with the aqueous RNP solution at a high total flow rate (≥ 12 mL/min) to form stable RNP-LNPs.
- Purification and Characterization: Dialyze against DPBS. Characterize particles for size, PDI, and encapsulation efficiency. Critical Note: Use a microfluidics-based method specifically optimized for proteins to avoid denaturation. [33]
→ Diagram: CRISPR RNP-LNP Assembly Workflow
The Scientist's Toolkit: Essential Reagents for LNP Research
Table 3: Key Research Reagent Solutions for LNP Development
| Reagent / Material | Function in LNP Formulation | Examples & Notes |
|---|---|---|
| Ionizable Lipids | Core structural component; enables encapsulation and endosomal escape. | ALC-0315 (Comirnaty), SM-102 (Spikevax). Newer thermostable variants support RNP delivery. [27] [33] [29] |
| PEGylated Lipids | Stabilizes particle size; reduces aggregation; modulates PK/PD. | DMG-PEG2000, DSPE-PEG2000. Molar ratio critical for controlling particle size and in vivo performance. [27] [29] |
| Structural Lipids | Enhances LNP bilayer stability and integrity. | DSPC, DOPE. Helps facilitate fusion with endosomal membranes. [29] |
| Cholesterol | Enhances stability and fluidity of the LNP membrane. | Molecular "filler" that improves packing and resilience in serum. [29] |
| SORT Molecules | Enables targeted delivery to specific tissues beyond the liver. | Adding a quaternary ammonium (cationic) lipid can redirect LNPs to the lungs. [33] [29] |
| Nucleic Acid Payloads | The therapeutic cargo. | siRNA: Chemically modified for stability. [31] sgRNA: Synthetic, high-purity. Cas9 mRNA/RNP: Codon-optimized mRNA or purified protein. [33] [29] |
LNPs have proven to be a transformative platform, enabling the clinical success of siRNA therapeutics and paving the way for the next generation of CRISPR-based gene therapies. The evolution from siRNA to RNP delivery, as demonstrated by recent breakthroughs in lung and liver editing, highlights the adaptability and power of LNP technology. [33] Future developments will focus on overcoming remaining challenges, including enhancing delivery to extrahepatic tissues through advanced targeting strategies (e.g., SORT molecules, ligand conjugation), improving the scalability and cost-effectiveness of manufacturing, and thoroughly understanding long-term safety profiles. [27] [34] [28] As the field progresses, the integration of AI-driven design for novel lipids and LNP formulations promises to further accelerate the development of precise and personalized genetic medicines. [30] [35] The protocols and insights provided here offer a foundation for researchers to leverage LNPs in their pursuit of novel genetic therapeutics.
Polymer-based vectors represent a promising class of non-viral delivery systems for genetic material, offering a compelling alternative to viral vectors by potentially mitigating immunogenicity concerns while providing tunable physicochemical properties. These vectors are engineered to navigate the complex intracellular environment, effectively encapsulating and protecting nucleic acids such as plasmid DNA, mRNA, and siRNA, and facilitating their delivery into target cells. The fundamental challenge in their design lies in optimizing two often competing characteristics: high transfection efficiency and favorable biocompatibility. Achieving this balance requires meticulous molecular engineering of polymer structures to control their interactions with biological systems, from cellular membranes to intracellular compartments.
The versatility of synthetic polymers allows for systematic modifications to their backbone, side chains, and functional groups, enabling fine-tuning of critical parameters including molecular weight, charge density, hydrophobicity, and degradation kinetics. Within the broader context of non-viral nanoparticle vectors research, polymer-based systems stand out for their scalability, reproducibility, and capacity for functionalization with targeting ligands. This application note provides a detailed technical overview of recent advances in polymer vector design, quantitative performance data, and standardized protocols to support researchers in developing next-generation gene delivery systems for therapeutic applications.
Quantitative Performance of Advanced Polymer Vectors
Recent research has yielded significant improvements in polymer vector performance, with novel materials demonstrating enhanced transfection efficiency and reduced cytotoxicity. The following tables summarize key quantitative data from cutting-edge studies for easy comparison of vector characteristics.
Table 1: Transfection Efficiency and Cytotoxicity of Featured Polymer Vectors
| Polymer Vector | Nucleic Acid Delivered | Cell Line / Model | Transfection Efficiency | Cytotoxicity (Cell Viability) | Key Structural Feature | Citation |
|---|---|---|---|---|---|---|
| DP50-PE6 (POx) | mRNA (Fluc) | 293T cells (in vitro) | 3.3 × 105-fold increase vs. parent polymer | Maintained high viability | PAmOx backbone with C10 alkyl chains from 1,2-epoxydecane | [36] |
| DP50-PE6 (POx) | mRNA (OVA) | B16-OVA melanoma (in vivo) | >90% tumor suppression (with anti-PD1) | Good biocompatibility observed | Spleen-targeting after IV administration | [36] |
| PBAE Nanocarriers | Plasmid DNA | Jurkat T cells | Up to 37% transfection | Minimal cytotoxicity | Low molecular weight, biodegradable | [37] |
| PBAE Nanocarriers | Plasmid DNA | Primary T cells | ~5% transfection | Minimal cytotoxicity | Optimized DNA-to-polymer ratio | [37] |
| OM-pBAE (Coated AAV) | Transgene for DMD | In vitro & in vivo DMD model | Superior transduction efficiency | Improved protection vs. neutralizing antibodies | Polymer coating evades immune response | [38] |
| STAR-CXP (Polyaminoacid) | pDNA, siRNA, saRNA | Comparative in vitro | Up to 9× higher than jetPEI | Reduced immunogenicity in human serum | Biodegradable, nuclear localization | [39] |
Table 2: Physicochemical Properties and Formulation Parameters
| Polymer Vector | Typical Formulation N/P Ratio | Polyplex/Particle Size (nm) | Zeta Potential (mV) | Key Administration Routes Tested | Optimal DP / Mw |
|---|---|---|---|---|---|
| DP50-PE6 (POx) | ~30/1 (mass ratio) | Not specified | Not specified | Intravenous, Intramuscular | DP 50 |
| PBAE Nanocarriers | Varied DNA-to-polymer ratios | Characterized for size and dispersion | Characterized for surface charge | Ex vivo T cell transfection | Low Mw |
| STAR-CXP | System-dependent | Stable nanoparticles, resists aggregation | Reduced surface charge with PSar shielding | Intravenous, Intramuscular | Not specified |
| PEI (Benchmark) | System-dependent | Varies with formulation | Highly positive (↓ with shielding) | Intratumoral, Intramuscular (non-systemic) | Broad Mw ranges |
The data in Table 1 highlights remarkable achievements in vector performance. The POx-based vector DP50-PE6 demonstrates an extraordinary 330,000-fold enhancement in mRNA transfection efficiency over its parent polymer, underscoring the profound impact of strategic hydrophobic modification [36]. Similarly, PBAE nanocarriers achieve notable transfection in hard-to-transfect primary T cells, a critical milestone for cell-based immunotherapies [37]. The >90% tumor suppression rate achieved by DP50-PE6 in a melanoma model, combined with the spleen-targeting capability observed after intravenous administration, positions this polymer as a particularly promising platform for mRNA vaccines and cancer immunotherapies [36].
Detailed Experimental Protocols
Protocol: Formulation of POx-Based Polyplexes for mRNA Delivery
This protocol outlines the synthesis of amino-functionalized poly(2-oxazoline) (POx) vectors and their complexation with mRNA, based on the highly effective DP50-PE6 polymer [36].
Reagents and Materials:
- Poly[2-(5-aminopentyl)-2-oxazoline] (PAmOx50, DP = 50)
- 1,2-epoxydecane (E6)
- Anhydrous Dimethyl Sulfoxide (DMSO) or Tetrahydrofuran (THF)
- mRNA of interest (e.g., encoding Firefly Luciferase or antigen)
- Nuclease-free water
- 1x Phosphate Buffered Saline (PBS), pH 7.4
- Equipment: Schlenk flask, Magnetic stirrer, Purification system (dialysis or SEC), Heated water bath, Vortex mixer, Dynamic Light Scattering (DLS) instrument.
Procedure:
- Polymer Synthesis (DP50-PE6): a. Reaction Setup: In a Schlenk flask under an inert atmosphere, dissolve PAmOx50 (1.0 g, ~20 µmol) in 10 mL of anhydrous DMSO. b. Epoxide Modification: Add 1,2-epoxydecane (E6, 10 mol equivalent per amine group) to the reaction mixture. c. Polymer Modification: Stir the reaction mixture at 60°C for 48 hours to allow for the ring-opening reaction between the primary amines of PAmOx and the epoxide groups. d. Purification: Purify the resulting DP50-PE6 polymer by dialysis against ethanol or methanol for 24 hours, followed by lyophilization. Alternatively, use size exclusion chromatography. e. Characterization: Confirm the chemical structure and grafting efficiency by ¹H NMR spectroscopy [36].
- Polyplex Formation (Complexation): a. Polymer Solution: Prepare a stock solution of DP50-PE6 in nuclease-free water or PBS at a concentration of 1 mg/mL. Filter sterilize (0.22 µm). b. mRNA Solution: Dilute the mRNA to a working concentration of 0.05 mg/mL in nuclease-free PBS. c. Complexation: Add the desired volume of the polymer solution to an equal volume of the mRNA solution to achieve a polymer-to-mRNA mass ratio of 30:1. For example, mix 30 µL of polymer stock (1 mg/mL) with 1 µL of mRNA stock (0.05 mg/mL) and 29 µL of PBS. Gently vortex the mixture for 3-5 seconds. d. Incubation: Allow the polyplexes to form by incubating the mixture at room temperature for 20-30 minutes. The solution may turn slightly opaque. e. Quality Control: Characterize the resulting polyplexes for size (hydrodynamic diameter) and surface charge (zeta potential) using Dynamic Light Scattering (DLS) prior to use.
Protocol: Transfection of Adherent Cells (293T) with POx/mRNA Polyplexes
This protocol describes the in vitro transfection of human embryonic kidney (293T) cells to evaluate the performance of the formulated polyplexes [36].
Reagents and Materials:
- DP50-PE6/mRNA polyplexes (from Protocol 3.1)
- 293T cell line
- Complete growth medium (e.g., DMEM with 10% FBS)
- Opti-MEM or serum-free medium
- Trypsin-EDTA solution
- 96-well or 24-well tissue culture plates
- Equipment: Cell culture incubator (37°C, 5% CO₂), Laminar flow hood, Centrifuge, Microplate reader or luminescence detector.
Procedure:
- Cell Seeding: Seed 293T cells in a 96-well plate at a density of 1.0 x 10⁴ cells per well in 100 µL of complete growth medium. Incubate the plate for 18-24 hours at 37°C in a 5% CO₂ incubator until the cells reach 60-80% confluency.
- Transfection Medium Exchange: Before transfection, carefully aspirate the growth medium and wash the cell monolayer once with PBS. Replace the medium with 100 µL of Opti-MEM or serum-free medium.
- Polyplex Application: Add the pre-formed DP50-PE6/mRNA polyplexes (e.g., 10 µL per well from a 60 µL total preparation) directly to the cells in serum-free medium. Gently swirl the plate to ensure even distribution.
- Incubation and Expression: Incubate the cells with the polyplexes for 4-6 hours at 37°C in a 5% CO₂ incubator.
- Post-Transfection Medium Exchange: After the incubation period, carefully aspirate the transfection mixture and replace it with 100 µL of fresh complete growth medium. Continue to incubate the cells for a further 24-48 hours to allow for protein expression.
- Efficiency Analysis: a. Luciferase Expression: If the mRNA encodes luciferase, lyse the cells using a passive lysis buffer and measure luminescence intensity using a microplate reader according to the luciferase assay system manufacturer's instructions. Normalize the relative light units (RLU) to the total protein content in the lysate (RLU/mg protein) [36]. b. Viability Assessment: Perform a parallel MTT or Alamar Blue assay to assess cell viability 24 hours post-transfection to ensure biocompatibility.
Protocol: T Cell Transfection using PBAE Nanocarriers
This protocol details the use of biodegradable Poly(β-amino ester) (PBAE) nanocarriers for transfecting Jurkat and primary T cells, a key step in cell-based cancer immunotherapy [37].
Reagents and Materials:
- Low molecular weight PBAE polymer (synthesized via Michael addition)
- Plasmid DNA (e.g., encoding GFP or CAR construct)
- Jurkat T cell line or isolated primary human T cells
- RPMI-1640 medium supplemented with 10% FBS and GlutaMax
- Interleukin-2 (IL-2) for primary T cell culture
- Anti-CD3/CD28 antibodies for T cell activation
- Opti-MEM medium
- Equipment: Flow cytometer, Confocal microscope, Cell culture incubator.
Procedure:
- Nanocarrier Formulation: a. Polymer Solution: Dissolve PBAE polymer in DMSO at 100 mg/mL, then dilute in acetate buffer (pH 5.0) to a final concentration of 1-5 mg/mL. b. DNA Solution: Dilute plasmid DNA in the same acetate buffer. c. Complexation: Rapidly mix the PBAE solution with the DNA solution at various DNA-to-polymer mass ratios (e.g., 1:10 to 1:50). Vortex for 30 seconds. d. Incubation: Incubate the mixture at room temperature for 15-30 minutes to allow for nanocarrier self-assembly. e. Characterization: Measure the size, polydispersity index (PDI), and zeta potential of the nanocarriers using DLS [37].
T Cell Preparation and Transfection: a. Cell Culture: Maintain Jurkat cells or isolated primary T cells in RPMI-1640 complete medium. For primary T cells, activate with anti-CD3/CD28 antibodies and add IL-2 (e.g., 100 IU/mL) 48 hours prior to transfection. b. Transfection: Harvest the cells, count them, and resuspend them in Opti-MEM at a density of 1-2 x 10⁶ cells/mL. c. Nanocarrier Application: Add the formulated PBAE nanocarriers to the cell suspension. Use a DNA mass of 1-2 µg per 1 x 10⁶ cells. d. Incubation: Incubate the cell-nanocarrier mixture for 4-6 hours at 37°C. e. Recovery: Centrifuge the cells to remove the transfection mixture, resuspend them in fresh complete medium (with IL-2 for primary cells), and continue culture for 24-72 hours.
Efficiency and Viability Assessment: a. Flow Cytometry: Analyze transfection efficiency by measuring the percentage of GFP-positive cells using flow cytometry 24-48 hours post-transfection. b. Viability: Assess cell viability simultaneously using a flow cytometry-based assay (e.g., propidium iodide exclusion) or a metabolic assay like MTT.
Visualization of Polymer Vector Design and Workflow
The strategic design of polymer vectors involves creating a molecular structure that can navigate each step of the intracellular delivery pathway. The following diagrams, generated using DOT language, illustrate the core design logic and a standard experimental workflow.
Polymer Vector Design Logic
Diagram 1: Logic of functional polymer vector design. Strategic modifications to a polymer backbone impart specific functionalities that address each barrier to efficient and safe gene delivery, ultimately achieving the goal of balanced performance. POx: Poly(2-oxazoline); PBAE: Poly(β-amino ester); PEI: Polyethylenimine; PSar: Polysarcosine.
Experimental Workflow for Evaluation
Diagram 2: Key stages of polymer vector development. The workflow progresses linearly from synthesis to in vivo evaluation, with critical quantitative analyses performed at each stage to inform iterative design improvements. DLS: Dynamic Light Scattering; PDI: Polydispersity Index.
The Scientist's Toolkit: Research Reagent Solutions
Successful development and testing of polymer-based gene delivery systems require a suite of specialized reagents and materials. The following table details essential components for a research program in this field.
Table 3: Essential Research Reagents and Materials for Polymer-Based Gene Delivery
| Reagent/Material | Function/Purpose | Specific Examples & Notes |
|---|---|---|
| Cationic Polymers | Forms core of delivery vector; condenses nucleic acids via electrostatic interactions. | PAmOx (starting material for POx vectors) [36], PBAE (biodegradable) [37], PEI (benchmark, high cytotoxicity) [39]. |
| Hydrophobic Modifiers | Enhances membrane interaction and promotes endosomal escape, boosting transfection. | 1,2-epoxydecane (E6) for modifying POx amines [36]. Other alkyl epoxides or acrylates. |
| Shielding Polymers | "Stealth" coating to reduce polyplex charge, prevent aggregation, and lower immunogenicity. | Polysarcosine (PSar) [39], Polyethylene Glycol (PEG), Poly(2-oxazoline). |
| Nucleic Acid Cargos | The therapeutic or reporter genetic material to be delivered. | mRNA (e.g., Fluc, OVA) [36], Plasmid DNA (e.g., encoding GFP, CAR) [37], siRNA. |
| Formulation Buffers | Medium for polyplex self-assembly; pH and ionic strength critically impact particle properties. | Acetate Buffer (pH 5.0) for PBAE nanocarriers [37], PBS (pH 7.4), nuclease-free water. |
| Characterization Tools | To measure key physicochemical properties of the formulated nanocarriers. | DLS/Zeta Potential Analyzer for size and surface charge [37]. ¹H NMR for polymer structure confirmation [36]. |
| Cell Culture & Assays | Biological systems and tools to evaluate transfection performance and safety. | Cell Lines (e.g., 293T [36], Jurkat [37]). Primary T cells [37]. Luciferase Assay Kit, Flow Cytometer, MTT/XTT Viability Assay. |
The field of gene therapy is rapidly evolving, offering promising strategies for treating genetic disorders, cancers, and infectious diseases by introducing, silencing, or editing therapeutic genes. A significant challenge in this domain is developing safe and efficient vectors for delivering genetic materials such as DNA, mRNA, siRNA, and miRNA into target cells. While viral vectors demonstrate high transfection efficiency, their clinical application faces substantial hurdles including immunogenicity, insertional mutagenesis risks, limited gene cargo capacity, and complex manufacturing processes [40] [8]. Inorganic nanoparticles have emerged as promising non-viral vectors that can effectively overcome these limitations.
Gold, silica, and carbon-based nanoparticles offer distinct advantages for gene delivery applications, including superior safety profiles, scalability for manufacturing, structural and functional reconfigurability, and the ability to accommodate various sizes of genetic cargo [8] [17]. Their tunable physicochemical properties, ease of functionalization, and excellent biocompatibility make them particularly valuable for creating targeted delivery systems that can navigate biological barriers and efficiently transport genetic payloads to specific cells and even subcellular compartments [41] [42]. This application note provides a comprehensive overview of the current advances and experimental protocols for utilizing these inorganic nanoparticles in gene delivery systems, framed within the broader context of non-viral vector research.
Gold Nanoparticles (AuNPs) in Gene Delivery
Application Notes
Gold nanoparticles have gained significant attention in biomedical applications due to their unique properties, including surface plasmon resonance, high surface-area-to-volume ratio, tunable size and shape, and ease of functionalization [41]. In gene delivery, AuNPs serve as versatile nanocarriers that can be engineered to overcome multiple biological barriers.
Key Applications:
- Targeted Drug/Gene Delivery: AuNPs can be functionalized with specific ligands to deliver genetic materials straight to sick cells, reducing side effects and improving treatment efficacy. Their surfaces can be engineered with smart polymers, oligonucleotides, or cleavable linkers that enable precise, on-demand drug release in response to internal or external stimuli such as pH, temperature, or near-infrared light [41].
- Medical Imaging and Diagnostics: The plasmonic properties of AuNPs enable enhanced contrast in various imaging modalities, allowing for simultaneous diagnosis and treatment monitoring. They help medical imaging machines show clearer pictures, which is particularly valuable for early disease detection [43] [41].
- Photothermal Therapy: AuNPs can convert light to heat, enabling localized thermal ablation of diseased cells when combined with therapeutic genes [41].
- Immunotherapy Enhancement: Scientists are using nanogold to strengthen immunotherapy approaches, including creating vaccines that use gold particles to better train the immune system [43].
Mechanisms of Action: The gene delivery process using AuNPs involves multiple critical stages: (1) passive targeting and accumulation in tumor tissue via the enhanced permeability and retention (EPR) effect; (2) active targeting of specific cells through surface-modified ligands; (3) cellular internalization primarily through endocytosis pathways; and (4) intracellular trafficking and release of genetic payloads [41] [42]. The surface of AuNPs can be modified with specific biomolecules like antibodies or targeting ligands to enable selective targeting of cells that overexpress complementary receptor proteins, facilitating receptor-mediated endocytosis [41].
Table 1: Key Properties and Applications of Gold Nanoparticles in Gene Delivery
| Property | Description | Application in Gene Delivery |
|---|---|---|
| Surface Plasmon Resonance | Collective oscillation of electrons at surface | Enhanced imaging, photothermal therapy |
| High Surface Area | Large surface area to volume ratio | High loading capacity for genetic materials |
| Tunable Size & Shape | Sizes from 1-100 nm, various geometries | Optimized cellular uptake and biodistribution |
| Easy Functionalization | Surface modification with biomolecules | Targeted delivery, enhanced biocompatibility |
| Biocompatibility | Low toxicity, suitable for biological use | Reduced side effects, clinical suitability |
Experimental Protocols
Protocol 1: Green Synthesis of Gold Nanoparticles Using Plant Extracts
Principle: This protocol utilizes plant-derived phytochemicals as reducing and stabilizing agents for eco-friendly AuNPs synthesis, offering advantages over traditional chemical methods that often involve toxic reagents [44].
Materials:
- Gold salt precursor (Chloroauric acid, HAuCl₄)
- Plant materials (green tea, aloe vera, cinnamon, or turmeric)
- Deionized water
- Ethanol
- Centrifuge
- UV-Vis spectrophotometer
- Transmission Electron Microscope (TEM)
Procedure:
- Plant Extract Preparation: Wash and dry selected plant materials. Grind into fine powder. Prepare 10% (w/v) aqueous extract by boiling 10g of plant material in 100mL deionized water for 10 minutes. Filter through Whatman No. 1 filter paper.
- Gold Nanoparticle Synthesis: Prepare 1mM aqueous HAuCl₄ solution. Mix plant extract with gold solution in 1:9 ratio (v/v) under constant stirring at room temperature. Observe color change from pale yellow to ruby red, indicating nanoparticle formation.
- Purification: Centrifuge the solution at 15,000 rpm for 20 minutes. Discard supernatant and resuspend pellet in deionized water. Repeat three times.
- Characterization: Confirm synthesis by UV-Vis spectroscopy (peak at 520-580 nm). Determine size and morphology using TEM. Measure surface charge via zeta potential analysis.
Protocol 2: Functionalization of AuNPs for Gene Delivery
Principle: This protocol describes surface modification of AuNPs with cationic polymers and targeting ligands to enhance gene binding, cellular uptake, and targeted delivery [41].
Materials:
- Synthesized AuNPs
- Polyethyleneimine (PEI) or poly-L-lysine (PLL)
- N-Hydroxysuccinimide (NHS) and 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)
- Targeting ligands (folic acid, peptides, or antibodies)
- Genetic material (DNA, siRNA, miRNA)
Procedure:
- Surface Activation: Adjust AuNPs suspension to pH 7.4. Add EDC and NHS to final concentrations of 5mM and 2mM respectively. Incubate with gentle shaking for 30 minutes.
- Polymer Coating: Add PEI or PLL at optimal weight ratio (typically 1:2 polymer:gold ratio). Incubate for 4 hours at room temperature with continuous stirring.
- Ligand Conjugation: Add targeting ligands to functionalized AuNPs. For folic acid conjugation, use molar ratio of 1:50 (FA:AuNPs). React for 12 hours at 4°C.
- Genetic Material Loading: Mix functionalized AuNPs with genetic material at optimal N/P ratio (typically 10:1 to 20:1). Incubate for 30 minutes at room temperature to form stable complexes.
- Quality Control: Verify complex formation by gel retardation assay. Determine particle size and zeta potential using dynamic light scattering. Assess encapsulation efficiency.
Silica Nanoparticles in Gene Delivery
Application Notes
Mesoporous silica nanoparticles (MSNs) have emerged as attractive drug delivery carriers due to their unique structural properties, including large surface area, controllable pore structure, high loading capacity, and excellent biocompatibility [45]. For gene delivery applications, MSNs offer significant advantages in protecting genetic materials and controlling their release.
Key Applications:
- Sustainable Gene Delivery Systems: Green synthesis approaches utilizing biowaste sources like rice husk, wheat husk, and horsetail plant offer eco-friendly alternatives to conventional silica sources, reducing environmental impact while maintaining functionality [45].
- Stimuli-Responsive Delivery: MSNs can be engineered with gatekeepers to provide controlled, stimuli-responsive release of genetic materials in response to pH, enzymes, or light [45].
- Targeted Therapy: Functionalization with targeting ligands enables cell-specific delivery, enhancing therapeutic efficacy while minimizing off-target effects [45].
- Combined Diagnosis and Therapy: The modular nature of MSNs allows integration of imaging agents with therapeutic genes for theranostic applications.
Recent Advances: Research has demonstrated that MSNs synthesized from rice husk and horsetail show particularly promising properties for biomedical applications, including high purity silica, well-defined mesoporosity, high surface area, and controlled pore sizes [45]. When evaluated under physiologically relevant conditions using microfluidic platforms that mimic blood circulation, these MSNs exhibited significantly enhanced cellular uptake compared to static conditions, emphasizing the importance of physiological flow in optimizing nanoparticle-based drug delivery systems [45].
Table 2: Characterization of Green-Synthesized Mesoporous Silica Nanoparticles from Various Biowaste Sources
| Biosource | Silica Purity | Surface Area (m²/g) | Pore Size (nm) | Gene Loading Efficiency |
|---|---|---|---|---|
| Rice Husk | High | 450-550 | 2.5-3.5 | 85-92% |
| Horsetail | High | 420-500 | 2.8-3.8 | 82-90% |
| Wheat Husk | Medium | 380-470 | 3.0-4.0 | 78-85% |
| Oat Husk | Medium | 350-430 | 3.2-4.2 | 75-83% |
| Wheat Stalk | Low-Medium | 320-400 | 3.5-4.5 | 70-80% |
Experimental Protocols
Protocol 3: Green Synthesis of Mesoporous Silica Nanoparticles from Rice Husk
Principle: This protocol describes an eco-friendly approach to synthesize MSNs from rice husk biowaste, utilizing the high silica content naturally present in agricultural byproducts [45].
Materials:
- Rice husk
- Sodium hydroxide (NaOH)
- Hydrochloric acid (HCl)
- Cetyltrimethylammonium bromide (CTAB)
- Ethanol
- Muffle furnace
- Autoclave
- Centrifuge
Procedure:
- Pre-treatment: Wash rice husk thoroughly with deionized water to remove dirt and debris. Dry at 90°C for 3 hours. Acid-treat with 1M HCl at 100°C for 3 hours with continuous stirring at 600 rpm to remove metallic impurities.
- Calcination: Filter acid-treated samples and rinse with distilled water until neutral pH. Calcine at 550°C for 4 hours with heating rate of 5°C/min to obtain white rice husk ash (RHA).
- Silica Extraction: Dissolve 1g RHA in 18mL of 2M NaOH at 80°C for 3.5 hours to prepare sodium silicate solution. Filter to remove insoluble residues.
- Nanoparticle Formation: Add sodium silicate solution to 100mL distilled water containing 4.8g CTAB (template agent). Adjust pH to 11.25 using HCl. Stir for 1 hour, then transfer to autoclave and heat at 100°C for 24 hours.
- Template Removal: Collect white precipitate by centrifugation. Wash with ethanol and water. Calcine at 550°C for 4 hours to remove CTAB template. Characterize using BET, TEM, and FTIR.
Protocol 4: Gene Loading and Functionalization of MSNs
Principle: This protocol describes the loading of genetic materials into MSNs and surface functionalization for targeted gene delivery [45].
Materials:
- Synthesized MSNs
- (3-Aminopropyl)triethoxysilane (APTES)
- Genetic material (DNA, siRNA)
- N-Hydroxysuccinimide (NHS)
- 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)
- Targeting ligands
Procedure:
- Surface Amination: Disperse MSNs in ethanol containing 2% APTES. Reflux for 12 hours at 70°C to introduce amine groups. Wash with ethanol and dry under vacuum.
- Genetic Material Loading: Prepare genetic material solution in suitable buffer. Add to aminated MSNs at weight ratio of 1:10 (genetic material:MSNs). Incubate for 24 hours at 4°C with gentle shaking. Separate loaded MSNs by centrifugation.
- Surface Functionalization: Activate targeting ligands with NHS/EDC in MES buffer (pH 6.0) for 30 minutes. Add to genetic material-loaded MSNs. React for 12 hours at 4°C.
- Characterization: Determine loading efficiency by measuring supernatant absorbance. Confirm functionalization by zeta potential and FTIR. Evaluate release profile in different pH buffers.
Carbon-Based Nanoparticles in Gene Delivery
Application Notes
Carbon-based nanoparticles, including carbon nanotubes (CNTs), graphene, carbon dots, and fullerenes, have shown great potential in gene delivery applications due to their unique structural, electronic, and mechanical properties [42] [46]. Their large surface area, needle-like shape that promotes cellular uptake, and ability to functionalize with various molecules make them excellent candidates for delivering genetic materials.
Key Applications:
- Nucleus-Targeted Delivery: Carbon nanoparticles can be engineered to target the cell nucleus, the ultimate target for most anticancer drugs, enabling more efficient gene therapy [42].
- Photothermal Therapy: CNTs exhibit strong absorption in near-infrared regions, making them promising materials for photothermal treatment when combined with gene therapy [46].
- Gene Delivery Platforms: Carbon-based materials serve as excellent carriers for various nucleic acids, including plasmid DNA, siRNA, and miRNA, protecting them from degradation and facilitating cellular uptake [47] [42].
- Theranostic Applications: The intrinsic fluorescence of certain carbon nanoparticles enables simultaneous imaging and therapy, allowing real-time monitoring of treatment efficacy [42].
Mechanisms of Action: Carbon nanoparticles utilize multiple pathways for cellular internalization and gene delivery. Their needle-like structure allows efficient penetration through cell membranes, and they can be functionalized with nuclear localization signals to achieve nucleus targeting [42] [46]. The process involves four critical stages: (I) passive targeting and accumulation in tumor tissue via the EPR effect; (II) active targeting of tumor cells through surface-modified ligands; (III) internalization through endocytosis or direct penetration; and (IV) intracellular trafficking and nucleus targeting [42].
Table 3: Comparison of Carbon-Based Nanoparticles for Gene Delivery Applications
| Nanoparticle Type | Size Range | Key Advantages | Gene Delivery Applications | Limitations |
|---|---|---|---|---|
| Carbon Nanotubes (CNTs) | 1-100 nm diameter | High aspect ratio, excellent cellular uptake, large surface area | siRNA delivery, gene silencing, photothermal combination therapy | Potential toxicity concerns, complex purification |
| Carbon Dots (CDs) | <10 nm | Excellent biocompatibility, fluorescence properties, easy functionalization | Plasmid DNA delivery, siRNA, dsRNA for plant and animal systems | Limited loading capacity for large genes |
| Graphene Oxide | 20-200 nm | Large surface area, oxygen functional groups for conjugation | DNA and RNA delivery, photothermal therapy | Potential aggregation in biological media |
| Mesoporous Carbon Nanospheres | 50-300 nm | Tunable pore size, high loading capacity | Controlled release of genetic materials, combination therapy | Complex synthesis procedures |
Experimental Protocols
Protocol 5: Preparation of Functionalized Carbon Nanotubes for Gene Delivery
Principle: This protocol describes the functionalization of carbon nanotubes to improve water solubility, reduce toxicity, and enable efficient gene loading and delivery [42] [46].
Materials:
- Pristine single-walled or multi-walled carbon nanotubes
- Sulfuric acid (H₂SO₄) and nitric acid (HNO₃)
- Polyethyleneimine (PEI)
- Genetic material (siRNA, DNA)
- Dialysis membrane
- Ultrasonicator
- Centrifuge
Procedure:
- Acid Treatment: Mix CNTs with 3:1 (v/v) mixture of H₂SO₄:HNO₃. Sonicate for 4 hours at 35-40°C to introduce carboxyl groups. Dilute with deionized water and filter through 0.22μm membrane. Wash until neutral pH.
- PEI Functionalization: Dissolve acid-treated CNTs in deionized water. Add PEI at weight ratio of 1:1 (CNTs:PEI). Sonicate for 30 minutes, then stir for 24 hours at room temperature.
- Purification: Dialyze against deionized water for 48 hours to remove unreacted PEI. Lyophilize to obtain PEI-functionalized CNTs.
- Gene Loading: Prepare genetic material solution in RNase-free buffer. Add to functionalized CNTs at optimal weight ratio (typically 1:20 for siRNA:CNTs). Vortex and incubate for 30 minutes at room temperature.
- Characterization: Confirm functionalization by FTIR and Raman spectroscopy. Determine size and morphology by TEM and SEM. Assess gene binding efficiency by gel electrophoresis.
Protocol 6: Nucleus-Targeted Gene Delivery Using Carbon Nanoparticles
Principle: This protocol describes the construction of nucleus-targeted carbon nanoparticle systems for enhanced gene delivery to the cell nucleus, utilizing nuclear localization signals (NLS) to overcome the nuclear membrane barrier [42].
Materials:
- Functionalized carbon nanoparticles
- Nuclear localization signal peptides (e.g., SV40 T-antigen NLS)
- Crosslinking reagents (SMCC, SPDP)
- Genetic material
- Cell culture reagents
- Confocal microscopy supplies
Procedure:
- Surface Activation: Suspend functionalized carbon nanoparticles in PBS. Add heterobifunctional crosslinker SMCC (final concentration 5mM). Incubate for 1 hour at room temperature. Remove excess crosslinker by gel filtration.
- NLS Conjugation: Dissolve NLS peptide in PBS. Add to activated nanoparticles at molar ratio of 50:1 (NLS:nanoparticle). React for 12 hours at 4°C with gentle mixing.
- Purification: Remove unconjugated NLS by dialysis against PBS for 24 hours. Characterize conjugation efficiency by HPLC and zeta potential measurement.
- Gene Loading: Incubate NLS-conjugated nanoparticles with genetic material at optimal N/P ratio. Incubate for 30 minutes to form complexes.
- Cellular Uptake and Nuclear Localization Assessment: Treat cells with nanoparticle-gene complexes. After incubation, fix cells and stain nuclei. Analyze cellular uptake and nuclear localization by confocal microscopy and flow cytometry.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Essential Research Reagents for Nanoparticle-Mediated Gene Delivery
| Reagent/Material | Function | Application Examples |
|---|---|---|
| Chloroauric acid (HAuCl₄) | Gold precursor for AuNPs synthesis | Seed-mediated growth of gold nanorods, spherical AuNPs |
| Cetyltrimethylammonium bromide (CTAB) | Template and stabilizing agent | Synthesis of mesoporous silica nanoparticles, gold nanorods |
| Polyethyleneimine (PEI) | Cationic polymer for gene complexation | Surface functionalization of AuNPs, CNTs, and silica nanoparticles |
| (3-Aminopropyl)triethoxysilane (APTES) | Silane coupling agent for surface amination | Functionalization of silica nanoparticles for gene binding |
| N-Hydroxysuccinimide (NHS) | Carboxyl group activator | Conjugation of targeting ligands to nanoparticle surfaces |
| 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) | Crosslinking agent | Covalent attachment of biomolecules to nanoparticles |
| Folic acid | Targeting ligand for cancer cells | Surface modification for targeted delivery to folate receptor-positive cells |
| Nuclear localization signals (NLS) | Peptides for nuclear targeting | Enhancing nuclear uptake of gene delivery systems |
| DSPE-PEG | Lipid-polymer conjugate for stealth coating | Improving circulation time and reducing opsonization |
Visualization of Nanoparticle-Mediated Gene Delivery Pathways
Inorganic nanoparticles—including gold, silica, and carbon-based materials—offer versatile platforms for advancing non-viral gene delivery systems. Their tunable physicochemical properties, functionalization flexibility, and demonstrated efficacy in preclinical studies position them as promising alternatives to viral vectors. The protocols and application notes provided in this document offer researchers comprehensive methodologies for synthesizing, functionalizing, and utilizing these nanoparticles in gene delivery applications. As the field progresses, focus must remain on addressing challenges related to long-term safety, biodegradation, manufacturing scalability, and regulatory approval to fully translate these promising technologies from laboratory research to clinical applications that benefit patients.
The field of gene therapy is at a pivotal juncture, with extracellular vesicles (EVs) emerging as a transformative platform for nucleic acid delivery. These natural lipid nanoparticles represent a promising alternative to viral vectors and synthetic nanoparticles, offering superior biocompatibility, low immunogenicity, and inherent targeting capabilities [48]. EVs are membrane-bound nanostructures (30-150 nm) secreted by virtually all cell types, playing crucial roles in intercellular communication through their cargo of proteins, lipids, and nucleic acids [49] [50]. Their endogenous origin enables them to circumvent many limitations associated with conventional gene delivery systems, including pre-existing immune recognition, cytotoxicity, and inefficient intracellular trafficking [48] [51]. As the therapeutic potential of gene editing technologies like CRISPR/Cas9 continues to expand, EVs provide a versatile delivery platform capable of transporting these large molecular complexes across biological barriers, including the blood-brain barrier, while protecting them from degradation [49] [52]. This application note details the latest methodologies and technical considerations for leveraging EV-based platforms in gene therapy research and development.
EV Engineering Strategies for Gene Delivery
Cargo Loading Methodologies
Loading therapeutic nucleic acids into EVs requires sophisticated engineering approaches that balance efficiency with preservation of vesicle integrity.
Table 1: Comparison of Major EV Loading Techniques
| Method | Mechanism | Optimal Cargo | Efficiency | Key Advantages | Major Limitations |
|---|---|---|---|---|---|
| Electroporation | Electrical field creates transient pores in membrane | siRNA, miRNA, CRISPR-Cas9 components [49] | Moderate to High | Maintains cargo bioactivity; wide applicability | Potential vesicle aggregation; cargo precipitation [48] |
| Sonication | Ultrasound disrupts membrane integrity | Chemotherapeutic drugs, proteins, nucleic acids [49] | High | Efficient for various cargo types | Potential membrane damage; compromised structural integrity |
| Transfection of Parent Cells | Genetic engineering of donor cells to load cargo during EV biogenesis | mRNA, plasmid DNA, engineered proteins [50] | Variable | Natural loading process; preserves EV integrity | Limited control over final cargo concentration |
| Incubation with Permeabilizers | Saponin or other agents increase membrane permeability | Small molecules, dyes, nucleotides | Low to Moderate | Simple protocol; minimal equipment | Low efficiency for large nucleic acids |
| Freeze-Thaw Cycles | Membrane disruption through ice crystal formation | Proteins, small RNAs | Low | Technically simple and accessible | Very low loading efficiency; vesicle fusion |
Surface Modification for Targeted Delivery
Engineering EV surfaces enhances their targeting specificity and therapeutic precision. Both pre-isolation and post-isolation modification strategies have been developed.
Pre-isolation (Genetic Engineering): Parent cells are genetically modified to express targeting ligands (peptides, antibody fragments, or receptors) on the EV surface. This approach leverages endogenous sorting mechanisms to incorporate targeting molecules during EV biogenesis [50]. For instance, donor cells can be engineered to express Lamp2b fusion proteins incorporating neuron-specific targeting peptides, resulting in EVs with enhanced blood-brain barrier penetration and neural cell specificity [49].
Post-isolation (Direct Modification): Isolated EVs are chemically or physically modified with targeting moieties. Click chemistry, hydrophobic insertion, and covalent conjugation enable the attachment of homing devices including aptamers, antibodies, and glycosylation patterns [50]. While this approach offers precise control over ligand density, it may compromise membrane integrity and requires extensive purification steps.
Quality Control and Analytical Characterization
Robust characterization of EV preparations is essential for reproducible research and clinical translation. The following parameters must be rigorously assessed.
Table 2: Essential Quality Control Parameters for EV-based Gene Delivery Systems
| Parameter | Analytical Methods | Target Specifications | Clinical Relevance |
|---|---|---|---|
| Size Distribution | Nanoparticle Tracking Analysis (NTA), Dynamic Light Scattering (DLS) | 30-150 nm; PDI < 0.2 [53] | Biodistribution, tissue penetration |
| Concentration | NTA, Tunable Resistive Pulse Sensing | >1×10^10 particles/mL for in vivo studies [53] | Dosing accuracy and reproducibility |
| Surface Marker Profile | Western Blot, Flow Cytometry, ELISA | Positive for CD9, CD63, CD81; negative for calnexin, GM130 [48] [54] | Identity, purity, and potency assessment |
| Nucleic Acid Loading Efficiency | Fluorometric assays, qRT-PCR, PAGE | >50% encapsulation efficiency; protected from RNase degradation [48] | Therapeutic payload delivery capacity |
| Morphology | Transmission Electron Microscopy (TEM) | Cup-shaped morphology; intact membrane [55] | Structural integrity and vesicle quality |
| Endotoxin & Contaminants | LAL assay, protein quantification | Endotoxin < 0.25 EU/mL; minimal protein contaminants [53] | Safety and immunogenicity profile |
Experimental Protocols
Protocol: CRISPR/Cas9 Loading via Electroporation
This protocol details the efficient encapsulation of CRISPR/Cas9 ribonucleoproteins (RNPs) into mammalian cell-derived EVs through optimized electroporation.
Materials & Reagents:
- Purified EVs (1×10^11 particles) from mesenchymal stem cells
- CRISPR/Cas9 RNP complex (20 µg)
- Electroporation buffer (sucrose-based, isotonic)
- 4-mm electroporation cuvettes
- DNase I (RNase-free)
- PD-10 desalting columns
- Dulbecco's Phosphate Buffered Saline (DPBS)
Procedure:
- EV Preparation: Isolate EVs from MSC conditioned media using differential ultracentrifugation (100,000 × g for 70 min) or size-exclusion chromatography. Resuspend EV pellet in electroporation buffer to concentration of 2×10^10 particles/mL.
- Complex Preparation: Combine EVs with CRISPR/Cas9 RNP complexes at 1:5 (w/w) ratio in electroporation buffer. Incubate 10 min at room temperature.
- Electroporation: Transfer mixture to pre-chilled electroporation cuvette. Apply electrical pulse (1000 μF, 150 V, ∞ resistance) using Gene Pulser Xcell system.
- Post-treatment: Immediately after pulsing, incubate cuvette on ice for 30 min. Add DNase I (5 U/μg DNA) to degrade external nucleic acids, incubating for 15 min at 37°C.
- Purification: Remove unencapsulated cargo using PD-10 desalting column equilibrated with DPBS. Concentrate using 100-kDa centrifugal filters.
- Quality Control: Verify loading efficiency via fluorescence quantification if using labeled RNPs. Assess EV integrity by TEM and NTA.
Technical Notes: Optimize voltage and capacitance for specific EV sources. Include controls with scrambled gRNA. Assess functional delivery in recipient cells using Surveyor or T7E1 mismatch assays [49] [51].
Protocol: Targeted EV Production via Parent Cell Engineering
This method describes the generation of targeted EVs through genetic engineering of parent cells to express homing ligands on the EV surface.
Materials & Reagents:
- HEK293T or MSC cells
- Lentiviral vector encoding targeting ligand (e.g., RVG-Lamp2b)
- Polybrene (8 μg/mL)
- Puromycin (1-2 μg/mL)
- Serum-free medium optimized for EV production
- EV purification filters (0.22 μm)
Procedure:
- Cell Engineering: Transduce parent cells with lentiviral vectors encoding the fusion construct at MOI 10-20 in presence of polybrene. Select stable pools with puromycin for 7-10 days.
- EV Production: Culture engineered cells in serum-free medium for 48h. Collect conditioned media and remove cells/debris by centrifugation (2000 × g, 20 min).
- EV Isolation: Concentrate EVs using tangential flow filtration (100-kDa cutoff) followed by size-exclusion chromatography (qEV columns).
- Validation: Confirm surface ligand presence by flow cytometry (after binding to streptavidin beads) or immunogold labeling TEM.
- Functional Testing: Evaluate targeting specificity using cell binding assays with target vs. non-target cells. Quantify gene delivery efficiency using reporter systems [50] [54].
Technical Notes: Include empty vector controls. Validate target receptor expression in recipient cells. Optimize transduction efficiency to avoid overexpression artifacts.
Pathway and Workflow Visualization
EV Engineering and Application Workflow
EV-Mediated Gene Delivery Intracellular Pathway
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for EV-based Gene Delivery Studies
| Reagent/Category | Specific Examples | Research Application | Technical Considerations |
|---|---|---|---|
| EV Source Cells | Mesenchymal Stem Cells (MSCs), HEK293T, Dendritic Cells | EV production; inherent tropism properties | MSC-EVs offer immunomodulation; HEK293T enable high-yield production [53] |
| Isolation Kits | Total Exosome Isolation Kit, qEV Size Exclusion Columns, TEI Reagent | Rapid EV purification from conditioned media or biofluids | Trade-offs between purity, yield, and functional preservation; SEC offers high purity [55] |
| Characterization Antibodies | Anti-CD63, CD81, CD9, TSG101, Calnexin (negative marker) | EV identification and purity assessment by Western blot, flow cytometry | Implement MISEV guidelines for minimal characterization standards [48] |
| Nucleic Acid Cargo | siRNA, miRNA, mRNA, CRISPR-Cas9 components, plasmid DNA | Therapeutic/genetic modifier payloads | Cargo size and structure significantly impact loading efficiency and biological activity [49] [51] |
| Tracking Dyes | PKH67, DiD, DIR, CFSE | EV tracking, biodistribution studies, cellular uptake quantification | Dye concentration optimization critical to avoid EV aggregation; include proper controls [50] |
| Targeting Ligands | RVG peptide, iRGD, GE11, Transferrin, Aptamers | Cell-specific targeting; enhanced therapeutic precision | Genetic engineering vs. post-isolation conjugation offer different advantages [50] [54] |
Exosomes and extracellular vesicles represent a paradigm shift in gene delivery platform technology, offering unique advantages over conventional viral and synthetic vectors. Their innate biological properties—including low immunogenicity, natural stability in circulation, and an inherent ability to traverse biological barriers—position them as ideal vehicles for therapeutic genetic cargo. The engineering strategies outlined herein enable researchers to overcome natural limitations and create targeted, efficient gene delivery systems with applications across diverse therapeutic areas including oncology, neurology, and regenerative medicine. As the field advances, standardization of isolation protocols, scaling of production processes, and rigorous safety profiling will be crucial for clinical translation. The integrated approaches presented in this application note provide a foundation for harnessing the full potential of EV-based platforms in next-generation gene therapy applications.
The field of gene therapy has undergone a transformative shift with the advent of non-viral nanoparticle vectors, which offer a safer and more versatile alternative to traditional viral delivery systems. Unlike viral vectors, which can pose risks such as immunogenicity, insertional mutagenesis, and limitations in cargo capacity, non-viral vectors provide distinct advantages including low immunogenicity, ease of synthesis and modification, large cargo capacity, and improved safety profiles [56] [57]. These benefits have accelerated the clinical translation of non-viral gene therapies, particularly with the emergence of lipid nanoparticles (LNPs) as a premier delivery platform for CRISPR-based therapeutics and other genetic medicines.
The clinical implementation of non-viral vectors represents a critical advancement for treating otherwise incurable genetic disorders, enabling precise genome editing with unprecedented accuracy and efficiency [58]. This progress is evidenced by the growing number of approved therapies and late-stage candidates utilizing non-viral delivery platforms. The following sections detail the most significant clinical success stories, provide detailed experimental protocols, and analyze the key factors driving the successful development of these revolutionary therapies.
Approved Non-Viral Gene Therapies
Casgevy (Exagamglogene Autotemcel)
Therapy Overview: Casgevy, developed by Vertex Pharmaceuticals and CRISPR Therapeutics, represents a landmark achievement as the first FDA-approved CRISPR-based medicine. This ex vivo gene therapy is approved for treating sickle cell disease (SCD) and transfusion-dependent beta thalassemia (TBT) [59] [60]. The therapy utilizes a non-viral delivery approach where patient-derived hematopoietic stem cells are genetically modified outside the body to produce elevated levels of fetal hemoglobin.
Mechanism of Action: The therapeutic approach involves using CRISPR-Cas9 to precisely edit the BCL11A gene, a key regulator that suppresses fetal hemoglobin production after birth. By disrupting this gene in autologous CD34+ hematopoietic stem and progenitor cells, the therapy reactitates fetal hemoglobin expression, which effectively compensates for the defective adult hemoglobin in SCD and TBT patients [60]. The editing components are typically delivered via electroporation, a non-viral physical method that temporarily disrupts cell membranes to allow intracellular entry of CRISPR machinery.
Clinical Efficacy: Clinical trials demonstrated profound and durable treatment effects. Patients with TBT achieved transfusion independence, while those with SCD experienced resolution of vaso-occlusive crises [59]. The therapy has shown sustained effectiveness in long-term follow-up, establishing it as a potentially curative one-time treatment for these inherited blood disorders.
Table 1: Key Clinical Trial Results for Casgevy
| Indicator | Sickle Cell Disease | Transfusion-Dependent Beta Thalassemia |
|---|---|---|
| Primary Endpoint | Resolution of vaso-occlusive crises | Transfusion independence |
| Efficacy Rate | >90% of patients free from crises for ≥12 months | >90% of patients achieved independence |
| Duration of Effect | Sustained up to 24 months in trials | Sustained up to 24 months in trials |
| FDA Approval Date | December 2023 | January 2024 |
Patisiran (Onpattro)
Therapy Overview: While not a gene editing therapy, patisiran represents a pioneering RNAi therapeutic that utilizes LNP technology for gene silencing. Approved for hereditary transthyretin-mediated amyloidosis, this therapy demonstrated the clinical viability of LNPs for systemic delivery of nucleic acid therapeutics [57].
Delivery System: Patisiran employs stable nucleic acid lipid particles (SNALP) technology, which encapsulates siRNA targeting mutant and wild-type transthyretin (TTR) mRNA. The LNPs are systemically administered and preferentially accumulate in hepatocytes, where they mediate degradation of TTR mRNA, reducing production of the amyloidogenic protein [57].
Clinical Impact: Phase III trials demonstrated significant improvement in neurological impairment and quality of life measures compared to placebo, establishing a new standard of care for this progressive fatal disease and validating LNP technology for systemic gene targeting applications.
Late-Stage Clinical Candidates
Intellia Therapeutics' In Vivo CRISPR Therapies
Intellia Therapeutics has pioneered the development of fully in vivo CRISPR-based therapies delivered via LNPs, representing the next frontier in non-viral gene editing.
NTLA-2001 for hATTR: Intellia's lead candidate, NTLA-2001, targets hereditary transthyretin amyloidosis (hATTR) with groundbreaking clinical results. This therapy utilizes LNPs containing CRISPR-Cas9 mRNA and guide RNA targeting the TTR gene in hepatocytes [59].
Table 2: Clinical Progress of NTLA-2001 for hATTR
| Trial Phase | Patient Population | Key Results | Next Milestones |
|---|---|---|---|
| Phase I | Patients with hATTR with neuropathy and cardiomyopathy | ~90% reduction in TTR protein levels sustained up to 24 months | Phase III initiated |
| Phase III | Global recruitment for cardiomyopathy patients (n=500+) | Placebo-controlled trial ongoing | Regulatory submission expected 2026-2027 |
The Phase I data published in the New England Journal of Medicine demonstrated rapid, deep, and durable reductions in TTR protein levels, with all 27 participants who reached two years of follow-up maintaining sustained response [59]. Functional and quality-of-life assessments showed stabilization or improvement of disease-related symptoms, representing a potential transformative one-time treatment for this progressive condition.
NTLA-2002 for HAE: Intellia's second program targets hereditary angioedema (HAE) using similar LNP technology to disrupt the KLKB1 gene, reducing plasma kallikrein activity. Phase I/II results reported in October 2024 showed an average of 86% reduction in kallikrein and significant reduction in HAE attacks, with 8 of 11 participants in the high-dose group remaining attack-free during the 16-week observation period [59].
ChristianaCare's CRISPR Therapy for Chemotherapy Resistance
Researchers at ChristianaCare's Gene Editing Institute have developed a novel CRISPR-based approach to reverse chemotherapy resistance in lung cancer, demonstrating the expanding applications of non-viral gene editing beyond monogenic diseases.
Therapeutic Approach: This innovative strategy targets the NRF2 gene, a master regulator of cellular stress responses that drives chemotherapy resistance when overactive. Using CRISPR-Cas9 delivered via LNPs, the therapy specifically knocks out the mutated NRF2 gene in tumor cells, restoring sensitivity to standard chemotherapy drugs like carboplatin and paclitaxel [61].
Key Findings: Published in Molecular Therapy Oncology in November 2024, the research demonstrated that disrupting NRF2 in just 20-40% of tumor cells was sufficient to improve chemotherapy response and slow tumor growth in animal models [61]. This "bystander effect" significantly enhances the therapeutic potential, as achieving complete editing of all tumor cells is clinically challenging. Sequencing confirmed high specificity for the mutated NRF2 gene with minimal off-target effects [61].
Clinical Implications: This approach represents a paradigm shift in cancer treatment by enhancing the efficacy of existing chemotherapies rather than developing entirely new drugs. The successful use of LNPs for tumor-specific delivery highlights the potential for non-viral vectors in oncology applications, with clinical trials expected to follow [61].
Detailed Experimental Protocols
LNP Formulation and CRISPR Delivery Protocol
The following protocol details the methodology for formulating LNPs containing CRISPR-Cas9 components and assessing their editing efficacy, based on techniques used in the cited clinical successes [59] [61].
Materials Required:
- Ionizable lipid: Key component for endosomal escape
- Helper lipids: DOPE, cholesterol, DSPC-MPEG (stability and stealth properties)
- CRISPR payload: Cas9 mRNA and single-guide RNA (sgRNA)
- Microfluidic device: For controlled nanoparticle formation
- Dialysis membranes: For buffer exchange and purification
- Cell lines: Relevant target cells (e.g., HepG2 for liver delivery)
- Animal models: Disease-relevant in vivo models
Step-by-Step Procedure:
Lipid Preparation:
- Dissolve ionizable lipid, DOPE, cholesterol, and DSPC-MPEG in ethanol at molar ratio 50:10:38.5:1.5
- Heat mixture to 60°C for 10 minutes with vortexing to ensure complete dissolution
Aqueous Phase Preparation:
- Dilute Cas9 mRNA and sgRNA in sodium acetate buffer (pH 4.0)
- Maintain nitrogen-to-phosphate (N:P) ratio of 6:1 for optimal encapsulation
- Keep aqueous phase at 4°C to minimize RNA degradation
LNP Formation:
- Utilize microfluidic device with total flow rate of 12 mL/min
- Mix lipid and aqueous phases at 3:1 volumetric ratio
- Collect effluent in dialysis cassette for buffer exchange
Purification and Characterization:
- Dialyze against PBS (pH 7.4) for 18 hours at 4°C
- Concentrate using centrifugal filters (100 kDa MWCO)
- Determine particle size (target: 70-100 nm) by dynamic light scattering
- Measure zeta potential (target: -5 to +5 mV) and encapsulation efficiency (>90%)
In Vitro Testing:
- Transfect target cells at varying doses (0.1-1.0 μg mRNA/well)
- Harvest cells 72 hours post-transfection for genomic DNA extraction
- Assess editing efficiency via T7E1 assay or next-generation sequencing
In Vivo Administration:
- Administer via tail vein injection in animal models
- Dose range: 1-3 mg CRISPR mRNA/kg body weight
- Monitor editing efficiency in target tissues over 2-4 weeks
Protocol for Assessing Tumor Chemosensitization
This protocol details the methodology for evaluating CRISPR-mediated reversal of chemotherapy resistance, based on the ChristianaCare approach [61].
Materials:
- Cancer cell lines: With documented chemotherapy resistance (e.g., NRF2 mutations)
- CRISPR-LNP formulation: Targeting resistance gene (NRF2)
- Chemotherapeutic agents: Standard-of-care drugs (carboplatin, paclitaxel)
- Cell viability assays: MTT or CellTiter-Glo
- Animal models: Immunocompromised mice for xenograft studies
Procedure:
In Vitro Sensitization Assessment:
- Seed cancer cells in 96-well plates (5,000 cells/well)
- Treat with CRISPR-LNPs at optimized concentration
- 48 hours post-transfection, add chemotherapeutic agents at IC50 concentration
- Incubate for additional 72 hours and assess viability
- Include controls: untreated, chemotherapy alone, CRISPR-LNPs alone
Gene Editing Confirmation:
- Extract genomic DNA from parallel samples
- Amplify target region by PCR and sequence
- Calculate indel percentage using TIDE analysis or similar tools
In Vivo Efficacy Studies:
- Establish tumor xenografts (1×10^6 cells/mouse)
- Randomize animals when tumors reach 150-200 mm³
- Administer CRISPR-LNPs intravenously (days 0, 7, 14)
- Initiate chemotherapy per standard dosing schedule
- Monitor tumor volume twice weekly for 4-6 weeks
Statistical Analysis:
- Compare tumor growth curves across treatment groups
- Calculate percentage of animals with tumor regression
- Perform histopathological analysis of harvested tumors
The Scientist's Toolkit: Essential Research Reagents
Successful development of non-viral gene therapies requires specialized reagents and materials optimized for nucleic acid delivery and gene editing applications.
Table 3: Essential Research Reagents for Non-Viral Gene Therapy Development
| Reagent Category | Specific Examples | Function & Application |
|---|---|---|
| Ionizable Lipids | DLin-MC3-DMA, SM-102, ALC-0315 | Enable encapsulation and endosomal escape of nucleic acids |
| Helper Lipids | DSPC, DOPE, Cholesterol | Stabilize LNP structure and enhance membrane fusion |
| PEGylated Lipids | DMG-PEG2000, DSG-PEG2000 | Provide stealth properties and prevent aggregation |
| CRISPR Components | Cas9 mRNA, sgRNA, RNP complexes | Active editing machinery for genetic modification |
| Microfluidic Devices | NanoAssemblr, staggered herringbone mixer | Enable reproducible, scalable LNP production |
| Analytical Instruments | DLS, NTA, HPLC | Characterize size, distribution, and encapsulation efficiency |
| Cell Lines | HepG2 (liver), HEK293 (kidney), primary cells | Model systems for testing delivery efficiency |
| Animal Models | C57BL/6 mice, non-human primates | Preclinical assessment of safety and efficacy |
The clinical success stories of approved therapies and late-stage candidates utilizing non-viral nanoparticle vectors underscore a fundamental transformation in gene therapy development. The progress from ex vivo applications like Casgevy to fully in vivo systemic delivery platforms such as Intellia's LNP-CRISPR therapies demonstrates the rapid advancement and expanding potential of non-viral delivery systems. These technologies have overcome previous limitations associated with viral vectors, including immunogenicity, cargo constraints, and manufacturing complexities.
The experimental protocols and research tools detailed in this document provide a framework for continued innovation in non-viral gene therapy development. As the field progresses, key challenges remain in optimizing tissue-specific delivery, enhancing editing efficiency, and ensuring long-term safety. However, the current clinical successes establish a robust foundation for the next generation of genetic medicines that will increasingly utilize non-viral vectors to treat a broadening spectrum of human diseases.
Solving the Delivery Puzzle: Strategies to Enhance Efficiency and Specificity
Overcoming Endosomal Degradation and Cytoplasmic Release
A central challenge in gene delivery using non-viral nanoparticle vectors is navigating the endosomal-lysosomal pathway. After cellular uptake, most nanocarriers become trapped within endosomes, which mature into lysosomes where the harsh acidic environment and potent hydrolytic enzymes degrade the therapeutic payload [62] [63]. Endosomal escape is the critical process where delivery vehicles must disrupt the endosomal membrane to release their genetic cargo into the cytoplasm for biological activity. This document details the primary mechanisms, quantitative challenges, and provides standardized protocols for designing and evaluating non-viral vectors capable of efficient endosomal escape and cytoplasmic release, framed within the broader research context of advancing gene delivery systems.
Mechanisms of Endosomal Escape
Non-viral vectors employ distinct biophysical mechanisms to facilitate endosomal escape. Understanding these is crucial for rational vector design.
- The Proton Sponge Effect: Cationic polymers with high buffering capacity, such as polyethyleneimine (PEI), absorb incoming protons as the endosome acidifies (pH drops from ~7.4 to ~5.0). This influx of protons is accompanied by chloride ions, leading to osmotic swelling, endosomal membrane rupture, and payload release into the cytosol [17].
- Lipid Membrane Disruption: Ionizable lipid nanoparticles (LNPs) are engineered to become positively charged in the acidic endosomal environment. This positive charge promotes interaction with and disruption of the anionic endosomal membrane, facilitating escape. The efficiency is highly dependent on the lipid's pKa, alkyl chain saturation, and branching, which influence the transition to fusogenic inverted hexagonal phases (HII) that destabilize the endosomal bilayer [63].
- Membrane Fusion and Pore Formation: Inspired by natural systems like extracellular vesicles (EVs), some synthetic systems utilize fusion peptides or lipids that directly merge with the endosomal membrane or form transient pores, allowing the therapeutic payload to exit [63].
The following diagram illustrates the primary pathways and barriers a non-viral vector encounters from cellular uptake to cytoplasmic release.
Quantitative Performance of Endosomal Escape Strategies
The efficiency of endosomal escape varies significantly between vector types. The table below summarizes key performance metrics for major classes of non-viral vectors, underscoring the need for improved designs.
Table 1: Performance Metrics of Non-Viral Vectors in Endosomal Escape
| Vector Type | Key Component | Escape Mechanism | Reported Escape Efficiency | Key Influencing Factors |
|---|---|---|---|---|
| Lipid Nanoparticles (LNPs) | Ionizable Lipids (e.g., DLin-MC3-DMA, SM-102) | Membrane Disruption / Phase Transition | ~1-4% of siRNA released from endosome [63] | pKa (~6-7), alkyl chain unsaturation, branching [63] |
| Cationic Polymers | Polyethyleneimine (PEI) | Proton Sponge Effect | Varies by polymer; can be superior to early LNPs | Molecular weight, branching, buffering capacity [62] [17] |
| Extracellular Vesicles (EVs) | Natural lipid & protein composition | Membrane Fusion / Native Processes | >10-fold higher than some commercial LNPs [63] | Cellular source, surface proteins, intrinsic properties [63] |
| Cell-Penetrating Peptides (CPPs) | TAT, PEN, R9 peptides | Various (Induced Endocytosis, Direct Translocation) | Dependent on sequence and cargo [64] | Peptide sequence, concentration, cargo type [64] |
Experimental Protocols
This section provides detailed methodologies for key experiments evaluating endosomal escape.
Protocol: Quantifying Endosomal Escape via Fluorophore-Based Assay
This protocol uses a double-quenched fluorescent probe that only emits signal upon lysosomal degradation and cytoplasmic release.
Research Reagent Solutions
Table 2: Essential Reagents for Fluorophore-Based Escape Assay
| Reagent | Function / Description | Example / Note |
|---|---|---|
| Ionizable Lipids | Core functional lipid for LNP formulation; enables endosomal disruption. | DLin-MC3-DMA, SM-102, ALC-0315 [63] |
| Helper Lipids | Stabilize LNP structure and can enhance fusogenicity. | DSPC, DOPE [63] [65] |
| PEGylated Lipids | Provide stealth properties, reduce opsonization; can inhibit uptake/escape (PEG Dilemma) [63]. | DMG-PEG, ALC-0159 |
| Cationic Polymers | Condense nucleic acids and facilitate escape via proton sponge effect. | Branched PEI, PAMAM dendrimers [62] [17] |
| Double-Quenched Fluorophore | Sensor probe (e.g., siRNA-Cy5) quenched by both internal quencher and endosomal environment; signal increases upon release/dequenching. | Useful for high-throughput screening [63] |
| Chloroquine / Bafilomycin A1 | Lysosomotropic agents; inhibit endosomal acidification and lysosomal function. Used as control to confirm escape pathway [64]. |
Methodology
- Vector Formulation: Formulate LNPs using microfluidic mixing. For a standard siRNA LNP, use a molar ratio of 50:10:38.5:1.5 (Ionizable Lipid:DSPC:Cholesterol:PEG-Lipid). Encapsulate a double-quenched Cy5-labeled siRNA probe [63].
- Cell Seeding and Transfection: Seed HeLa or HEK-293 cells in a 96-well black-walled plate at a density of 2.5 x 10^4 cells/well and culture for 24 hours.
- Treatment: Treat cells with the formulated vectors (e.g., 50 nM siRNA final concentration). Include controls: untreated cells, cells with free fluorescent siRNA (positive control for direct cytoplasmic signal), and cells pre-treated with 100 µM Chloroquine for 1 hour (escape enhancement control) [64].
- Imaging and Analysis: At 4, 8, 12, and 24 hours post-transfection, image cells using a high-content confocal microscope. Quantify fluorescence intensity (Cy5 channel) using image analysis software (e.g., ImageJ). Normalize the signal from the LNP-treated group to the positive and negative controls to calculate the relative escape efficiency.
Protocol: Evaluating Endocytic Pathways with Pharmacological Inhibitors
Understanding the cellular uptake pathway is critical as it influences subsequent endosomal trafficking and escape potential.
Methodology
- Cell Seeding: Seed appropriate cells (e.g., A549) in a 24-well plate and grow to 70-80% confluence.
- Inhibitor Pre-treatment: Pre-treat cells for 1 hour with specific endocytosis inhibitors dissolved in culture media [64]:
- Chlorpromazine (CPZ): 10 µg/mL to inhibit clathrin-mediated endocytosis (CME).
- Nystatin: 25 µg/mL to inhibit caveolae-mediated endocytosis (Cav).
- EIPA: 50 µM to inhibit macropinocytosis.
- Include a vehicle control (e.g., DMSO <0.1%).
- Vector Incubation: Add fluorescently labeled vectors (e.g., Cy3-labeled LNPs or CPPs) to the inhibitor-containing media and incubate for 2-4 hours.
- Analysis: Wash cells thoroughly with PBS and analyze cellular fluorescence via flow cytometry. A significant reduction in fluorescence in a specific inhibitor group indicates the primary uptake pathway involved [64].
The workflow for investigating the mechanism of endosomal escape, from inhibitor treatment to data interpretation, is outlined below.
The Scientist's Toolkit: Key Research Reagent Solutions
A curated list of essential materials and their functions for research in this field.
Table 3: Key Research Reagents for Endosomal Escape Studies
| Category | Reagent / Material | Primary Function in Research |
|---|---|---|
| Lipid Components | DLin-MC3-DMA, SM-102, ALC-0315 | Benchmark ionizable lipids for LNP formulation; study structure-activity relationships [63]. |
| DOPE (Dioleoylphosphatidylethanolamine) | Helper lipid that promotes transition to hexagonal HII phase, enhancing membrane fusion and escape [65]. | |
| DMG-PEG2000 | PEG-lipid used to control nanoparticle stability, circulation time, and cellular uptake (PEG Dilemma) [63]. | |
| Polymeric Vectors | Branched PEI (25 kDa) | "Gold standard" cationic polymer for studying the proton sponge effect; high transfection efficiency but with cytotoxicity concerns [62] [17]. |
| PAMAM Dendrimers | Highly branched, monodisperse polymers for gene delivery; customizable surface groups [62] [17]. | |
| Bioactive Molecules | Cell-Penetrating Peptides (CPPs: TAT, PEN, R9) | Promote cellular uptake and can be conjugated to cargo or vectors to study induced endocytosis pathways [64]. |
| Assay Kits & Probes | Double-Quenched Fluorogenic Probes (e.g., siRNA-Cy5) | Directly quantify endosomal escape efficiency via fluorescence dequenching upon cytoplasmic release [63]. |
| Lysotracker & pH-Sensitive Dyes | Track endosomal-lysosomal maturation and measure intra-organelle pH to correlate with vector escape kinetics. | |
| Pharmacological Inhibitors | Chloroquine, Bafilomycin A1 | Inhibit endosomal acidification and lysosomal function; used as controls to validate escape mechanisms [64]. |
| Chlorpromazine, Nystatin, EIPA | Elucidate the primary endocytic pathways involved in cellular uptake of vectors [64]. |
Achieving Tissue-Specific Targeting Beyond the Liver
The inherent liver tropism of non-viral nanoparticle vectors represents a fundamental challenge in expanding gene therapy applications to other tissues. This bias stems from the liver's physiological role in filtering circulating particulates and the natural affinity of conventional lipid nanoparticles (LNPs) for hepatic cells via apolipoprotein E (ApoE) opsonization and subsequent uptake through the very low-density lipoprotein receptor (VLDLR) [66]. While this property benefits liver-targeted therapies for inherited metabolic diseases, it creates a significant delivery barrier for treating conditions affecting other organs [15] [1]. Current organ-specific delivery systems predominantly enable targeted mRNA expression but fail to adequately resolve persistent hepatic accumulation, creating a desynchrony between nanoparticle distribution and therapeutic translation that limits clinical translation of precise mRNA drugs [67]. This application note details innovative strategies and methodologies to overcome biological barriers and achieve veritable tissue-specific targeting beyond the liver, enabling advanced gene therapies for pulmonary, cardiovascular, and other extrahepatic diseases.
Strategic Approaches for Extrahepatic Targeting
Core Strategies and Their Performance Metrics
Three primary engineering approaches have demonstrated significant potential for redirecting nanoparticles to extrahepatic tissues: surface modification, formulation optimization, and novel lipid design. The table below summarizes the key strategies, their mechanisms, and their validated therapeutic performance.
Table 1: Strategic Approaches for Extrahepatic Targeting
| Strategy | Mechanism of Action | Target Tissue/Cells | Therapeutic Payload | Efficacy Metrics |
|---|---|---|---|---|
| Surface Modification [15] | Attachment of antibodies, peptides, aptamers, or small molecule sugars to bind specific cellular receptors | Vascular endothelium, immune cells, pulmonary epithelium | siRNA, mRNA, CRISPR-Cas9 | Increased target cell uptake (2-5 fold); Reduced hepatic accumulation (30-60%) |
| Formulation Optimization [67] [68] | Adjusting LNP composition ratios; Selective removal of cholesterol and phospholipids | Lung endothelium and epithelium | mRNA | Simultaneous mRNA accumulation and translation in lung; 90% reduction in hepatic expression |
| Novel Ionizable Lipid Design [67] | Degradable ester-core based lipids with branched, single-tailed structures | Lung endothelial cells | mRNA | Superior endosomal escape (65% higher membrane fusion); sustained stability (30 days at 4°C) |
| PEG-Lipid Content Modulation [68] | Balancing nanoparticle stability and cellular uptake through PEG percentage variation | Spleen, bone marrow | mRNA | Bell-shaped efficacy curve; Optimal in vivo performance at 5% DMG-PEG2000 |
Key Signaling Pathways and Experimental Workflows
The following diagram illustrates the strategic decision-making workflow for selecting appropriate extrahepatic targeting approaches based on research objectives and experimental constraints.
Detailed Experimental Protocols
Protocol 1: Formulation-Driven Lung Targeting via Cholesterol-Free LNPs
This protocol describes the synthesis and characterization of cholesterol-free LNPs for achieving simultaneous mRNA accumulation and translation in lung tissue, based on breakthrough research demonstrating that cholesterol removal effectively prevents hepatic nanoparticle accumulation [67].
Materials Required:
- Ionizable lipid (e.g., 6Ac1-C12 with degradable ester core)
- PEG-lipid (DMG-PEG2000)
- Helper lipid (DOPE optional)
- Cholesterol (for control formulations only)
- mRNA payload (e.g., firefly luciferase or eGFP mRNA)
- Anhydrous ethanol
- Acetate buffer (200 mM, pH 5.4)
- Microfluidic device or T-junction mixer
Procedure:
- Lipid Stock Preparation: Prepare individual lipid stock solutions in anhydrous ethanol:
- Ionizable lipid: 100 mg/mL
- DMG-PEG2000: 50 mg/mL
- DOPE (if used): 25 mg/mL
Formulation Composition: Mix lipid stocks at the following molar ratios for lung-targeted LNPs:
- Ionizable lipid: DMG-PEG2000 = 15:2 (molar ratio)
- Optional: Include DOPE at 20 mol% if enhanced membrane fusion required
- Explicitly exclude cholesterol from the formulation
Nanoparticle Assembly:
- Use a microfluidic device with staggered herringbone mixer or a T-junction setup
- Set aqueous to organic flow rate ratio at 3:1
- Rapidly mix the lipid-ethanol phase with acetate buffer (pH 5.4)
- Maintain total flow rate at 12 mL/min for consistent particle size
Buffer Exchange and Characterization:
- Dialyze formulated LNPs against PBS (pH 7.4) for 24 hours at 4°C
- Filter sterilize using 0.22 μm polyethersulfone membrane
- Characterize particle size, PDI, and zeta potential using dynamic light scattering
- Determine mRNA encapsulation efficiency via RiboGreen assay
Validation Metrics:
- Size: 80-120 nm
- PDI: <0.2
- Encapsulation efficiency: >90%
- In vivo lung expression: >10^6 RLU/mg protein (luciferase)
- Hepatic expression: <10^4 RLU/mg protein
Protocol 2: PEG-Lipid Content Optimization for Tissue-Specific Delivery
This protocol systematically evaluates the effect of PEG-lipid content on LNP performance, enabling researchers to balance nanoparticle stability against cellular uptake efficiency for different target tissues [68].
Materials Required:
- Ionizable lipid (synthetic, e.g., tris(2-aminoethyl)amine derivative)
- Cholesterol
- DOPE (helper lipid)
- DMG-PEG2000
- mRNA encoding reporter protein (luciferase or eGFP)
- HeLa cells and DC2.4 dendritic cells
- Animal model (e.g., C57BL/6 mice)
Procedure:
- LNP Library Formulation:
- Prepare base formulation with fixed ionizable lipid/DOPE ratio (40:10 mol%)
- Systematically vary DMG-PEG2000 content from 0.1% to 10% molar ratio
- Adjust cholesterol content inversely to maintain total lipid concentration
- Use constant N/P ratio of 12.5:1 for mRNA encapsulation
In Vitro Screening:
- Seed HeLa and DC2.4 cells in 96-well plates at 10,000 cells/well
- Transfect with LNP formulations containing 100 ng mRNA/well
- Incubate for 24-48 hours at 37°C, 5% CO2
- Assess transfection efficiency via luciferase activity or flow cytometry for eGFP
- Evaluate cytotoxicity using CCK-8 assay
In Vivo Biodistribution:
- Administer LNPs intravenously to mice (0.5 mg mRNA/kg)
- Image luciferase expression at 4, 8, 24, and 48 hours post-injection
- Harvest organs at 24 hours for ex vivo imaging and qPCR analysis
- Quantify mRNA accumulation in lung, liver, spleen, and kidney
Expected Results:
- Bell-shaped efficacy curve with optimal in vitro transfection at 1.5% DMG-PEG2000
- Optimal in vivo performance at 5% DMG-PEG2000
- PEG content-dependent shift in organ biodistribution
- Inverse relationship between PEG content and hepatic accumulation
Protocol 3: Surface Functionalization for Active Tissue Targeting
This protocol describes the conjugation of targeting ligands to LNP surfaces for active targeting of specific cell receptors in extrahepatic tissues [15].
Materials Required:
- Pre-formed LNPs (containing maleimide-functionalized PEG-lipid)
- Thiolated targeting ligands (antibodies, peptides, or aptamers)
- Purified ApoE (for competitive binding studies)
- Size exclusion chromatography columns (Sephadex G-25)
- Tris(2-carboxyethyl)phosphine (TCEP) reducing agent
Procedure:
- Ligand Preparation:
- Reduce disulfide bonds in thiolated ligands with 10 mM TCEP
- Purify reduced ligands using desalting columns
- Confirm free thiol groups using Ellman's assay
Conjugation Reaction:
- Incubate maleimide-containing LNPs with thiolated ligands
- Use 2:1 molar ratio of ligand to maleimide groups
- React in PBS (pH 7.0-7.4) for 4 hours at room temperature
- Quench reaction with 10 mM cysteine
Purification and Characterization:
- Remove unconjugated ligands using size exclusion chromatography
- Verify conjugation efficiency with fluorescence labeling
- Assess binding specificity to target cells via flow cytometry
- Evaluate in vitro targeting in presence/absence of ApoE
Validation Methods:
- Ligand density quantification: 30-50 ligands per nanoparticle
- Cell-specific uptake: >3-fold increase vs. non-targeted LNPs
- Competitive inhibition: >70% reduction with receptor blockers
- In vivo targeting: >5-fold increase in target tissue vs. non-targeted controls
The Scientist's Toolkit: Key Research Reagents
Table 2: Essential Reagents for Extrahepatic Targeting Research
| Reagent Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Ionizable Lipids | 6Ac1-C12, DLin-MC3-DMA, SM-102 [67] | mRNA complexation and endosomal escape | Ester-core degradable lipids enhance safety; Branching affects efficacy |
| PEG-Lipids | DMG-PEG2000, DSPE-PEG, ALC-PEG [68] | Stability, circulation time, immunogenicity modulation | Short tails (C14) enhance cellular uptake; Long tails prolong circulation |
| Helper Lipids | DOPE, DSPC [15] [67] | Structural stability, membrane fusion facilitation | DOPE preferred for mRNA; DSPC for siRNA |
| Targeting Ligands | Antibodies, peptides, aptamers, GalNAc [15] [1] | Active cellular targeting | Ligand density critical for efficacy; Potential immunogenicity |
| Characterization Tools | Dynamic light scattering, RiboGreen assay, FRET probes [67] [68] | Size, PDI, encapsulation efficiency, endosomal escape | FRET probes quantify membrane fusion efficiency |
Technical Considerations and Troubleshooting
Critical Parameters for Success
The following diagram illustrates the relationship between LNP composition, their physicochemical properties, and the resulting biological behavior, highlighting key tunable parameters for extrahepatic targeting.
Optimization Guidelines:
- Ionizable Lipid Structure: Multi-branched, single-tailed lipids with ester cores demonstrate superior endosomal escape and biodegradability [67].
- PEG-Lipid Content: Balance between in vitro transfection (optimal at 1.5%) and in vivo performance (optimal at 5%) requires careful optimization for each application [68].
- Component Selection: Cholesterol removal is essential for reducing hepatic accumulation but may require compensation with structural lipids [67].
- Ligand Density: Optimal targeting requires 30-50 ligands per nanoparticle; excessive density causes steric hindrance and reduced uptake [15].
Troubleshooting Common Challenges
- Persistent Hepatic Accumulation: Implement cholesterol-free formulations and increase PEG content to 5-7%. Incorporate ApoE competitors during formulation.
- Poor Endosomal Escape: Select ionizable lipids with pKa values between 6.2-6.5. Incorporate DOPE as a helper lipid and validate membrane fusion using FRET assays [67].
- Rapid Clearance: Optimize PEG-lipid structure and content. Consider alternative surface coatings such as poloxamers or cell membrane derivatives.
- Batch-to-Batch Variability: Standardize microfluidic mixing parameters. Implement rigorous quality control for size, PDI, and encapsulation efficiency.
The protocols and strategies detailed in this application note provide a systematic framework for achieving tissue-specific targeting beyond the liver using non-viral nanoparticle vectors. By leveraging formulation optimization, surface engineering, and novel lipid designs, researchers can overcome the inherent hepatic tropism of conventional LNPs and expand the therapeutic potential of gene medicines to pulmonary, cardiovascular, and other extrahepatic tissues. The continued refinement of these approaches promises to accelerate the development of precise, effective gene therapies for a broad spectrum of diseases.
Strategies for Enhancing Cellular Uptake and Nuclear Entry
For gene therapy using non-viral nanoparticle vectors, the journey from cellular exterior to nuclear interior presents a series of formidable biological barriers. Overcoming these hurdles is critical for achieving efficient transgene expression. This application note details the primary strategies and quantitative principles for enhancing cellular uptake and nuclear entry, providing researchers with structured protocols and data to inform vector design and experimental planning.
The efficacy of non-viral gene delivery is often constrained by multiple extracellular and intracellular obstacles. These include enzymatic degradation in the bloodstream, electrostatic repulsion at the cell membrane, inefficient endosomal escape, and the final challenge of nuclear entry [69] [57]. Quantitative understanding of these bottlenecks, such as the finding that only 0.1% of an administered poly(beta-amino ester) (PBAE)/DNA polyplex dose is internalized by cells, is essential for rational vector development [20].
Quantitative Analysis of Intracellular Trafficking
A quantitative understanding of the mass transfer bottlenecks in gene delivery is a prerequisite for designing improved vectors. The following data, derived from studies using polymeric and liposomal systems, provides key benchmarks.
Table 1: Quantitative Rate Constants and Efficiencies for Non-Viral Vector Trafficking
| Process Stage | Quantitative Metric | Reported Value | Experimental System | Citation |
|---|---|---|---|---|
| Cellular Uptake | Uptake Rate Constant (k_cell) | 7.5 × 10⁻⁴ hr⁻¹ | PBAE/DNA polyplex in human primary glioblastoma cells | [20] |
| Percentage of Added Dose Internalized | 0.1% | PBAE/DNA polyplex in human primary glioblastoma cells | [20] | |
| Nuclear Entry | Nuclear Internalization Rate (k_ni) | 1.1 hr⁻¹ | PBAE/DNA polyplex (rate of internalization of nuclear-associated plasmid) | [20] |
| Efficiency of Internalized DNA Reaching Nucleus | 12% | PBAE/DNA polyplex in human primary glioblastoma cells | [20] | |
| Plasmid Degradation | Fast Degradation Rate Constant | 0.62 hr⁻¹ | PBAE/DNA polyplex (accounted for via qPCR) | [20] |
| Slow Degradation Rate Constant | 0.084 hr⁻¹ | PBAE/DNA polyplex (accounted for via qPCR) | [20] | |
| Nuclear Transcription | Intranuclear Gene Copies Needed for Expression | ~1000x more than adenovirus | Lipofectamine Plus (LFN) vs. Adenovirus (Ad) | [70] |
The data in Table 1 highlights that cellular uptake is a major rate-limiting step for the studied PBAE polymer, with a very low uptake rate constant and percentage of internalized dose [20]. Furthermore, while nuclear entry of internalized DNA can be relatively efficient, the ultimate barrier to successful gene expression often lies in inefficient nuclear transcription, where non-viral systems require orders of magnitude more intranuclear gene copies to achieve expression levels comparable to viral vectors [70].
Strategic Approaches for Enhanced Delivery
The quantitative bottlenecks can be overcome through intelligent engineering of the nanoparticle's physicochemical properties and the use of functional biological motifs.
Engineering Nanoparticle Physicochemical Properties
The size, surface charge, and composition of nanoparticles are critical parameters that directly influence their interaction with biological systems.
- Particle Size Optimization: Nanoparticles in the range of 60-100 nm are more readily endocytosed. Particles smaller than 50 nm are rapidly cleared by the kidneys, while those larger than 300 nm risk activating the immune response and are more difficult for cells to internalize [57] [16]. The primary pathway for uptake of these optimized particles is often receptor-mediated endocytosis [57].
- Surface Charge Tuning: A positive or neutral surface charge (positive zeta potential) enhances interaction with the negatively charged cell membrane. However, a balance is crucial; excessively high positive charge causes significant cytotoxicity and non-specific protein adsorption, while too low a charge leads to poor cellular uptake and nanoparticle instability. A zeta potential around +35 mV is often targeted [57] [16].
- Surface Functionalization: Decorating the nanoparticle surface with targeting ligands (e.g., antibodies, folic acid, transferrin, peptides) promotes specific binding to receptors on target cells, enhancing cellular uptake via receptor-mediated endocytosis and improving specificity [57] [71].
- Composition and Stability: The use of ionizable lipids (e.g., in LNPs) or cationic polymers (e.g., PEI, PBAEs) is fundamental for nucleic acid complexation. Incorporating stealth components like polyethylene glycol (PEG) creates a hydrophilic layer that minimizes non-specific interactions with serum proteins, thereby enhancing nanoparticle stability and circulation half-life [57] [16] [72].
Employing Functional Motifs for Intracellular Trafficking
Beyond general properties, specific functional motifs can be incorporated to overcome intracellular barriers.
- Enhancing Endosomal Escape: The "proton sponge" effect, exhibited by polymers like PEI and PBAEs that have high buffering capacity, is a key strategy to disrupt endosomes and release genetic cargo into the cytoplasm [20] [57]. Ionizable lipids in LNPs are also designed to become positively charged in the acidic endosomal environment, promoting endosomal membrane disruption and cargo release [16] [72].
- Facilitating Nuclear Entry: The use of Nuclear Localization Signals (NLS) is a primary method to enhance nuclear import. These peptide sequences can be conjugated to the nanoparticle or encoded within the plasmid DNA itself. Furthermore, incorporating a DNA Nuclear Targeting Sequence (DTS), such as the SV40 DTS, can exploit cellular transcription factors that possess their own NLS, effectively hijacking the natural nuclear import machinery [20].
The logical relationship between the key barriers and the engineering strategies designed to overcome them is summarized in the following diagram:
Detailed Experimental Protocols
Protocol: Quantifying Cellular and Nuclear Uptake Rates via Flow Cytometry
This protocol, adapted from a primary research study, provides a method to quantitatively track the journey of fluorescently labeled plasmid DNA from cellular uptake to nuclear entry, enabling the calculation of key rate constants [20].
1. Principle: This assay uses flow cytometry to quantitatively distinguish plasmids in the cytoplasm, associated with the nuclear envelope, and internalized within the nucleus over time. By converting fluorescence to plasmid counts and accounting for degradation, a four-compartment mass-action model is used to determine rate constants for cellular uptake (kcell), nuclear envelope association (kne), and nuclear internalization (k_ni) [20].
2. Reagents and Equipment:
- Polymeric Vector: e.g., Poly(beta-amino ester) PBAE 447, synthesized and fractionated to a specific molecular weight [20].
- Plasmid DNA: e.g., eGFP-N1 plasmid, conjugated to Cy3 dye using a labeling kit (e.g., Mirus Label IT Tracker Cy 3 Kit). Determine the nucleotide-to-dye ratio (N:D) via spectrophotometry [20].
- Cell Culture: Relevant cell line (e.g., human primary glioblastoma cells).
- Key Equipment: Flow cytometer, fluorescent plate reader, gel permeation chromatography (GPC) system for polymer fractionation, qPCR machine.
3. Procedure: Step 1: Vector and Plasmid Preparation
- Synthesize and characterize your non-viral vector (e.g., PBAE via Michael addition) [20].
- Fractionate the polymer using GPC to obtain a narrow, defined molecular weight distribution for reproducible results [20].
- Conjugate plasmid DNA with Cy3 dye according to the manufacturer's protocol. Purify the conjugated plasmid and mix with unconjugated plasmid at a defined ratio (e.g., 1:4) to prevent fluorescence quenching. Confirm the N:D ratio using the provided equation [20].
Step 2: Cell Transfection and Sampling
- Seed cells at a defined density and allow to adhere.
- Transfert cells with the vector/Cy3-pDNA polyplexes at optimized conditions.
- At multiple time points post-transfection (e.g., 1, 2, 4, 8, 12, 24 hours), harvest cells. For each time point, split the sample into three aliquots for different staining procedures [20].
Step 3: Staining for Compartmental Discrimination
- Aliquot A (Total Cellular DNA): Lyse cells completely to measure all internalized Cy3-pDNA. This represents the sum of Pcyto, Pne, and P_ni.
- Aliquot B (Cytosolic + Nuclear Envelope DNA): Permeabilize the cell membrane but not the nuclear membrane (e.g., with digitonin). This measures Pcyto and Pne.
- Aliquot C (Background/Control): Use to account for autofluorescence and non-specific staining.
Step 4: Flow Cytometry and Data Analysis
- Analyze all aliquots via flow cytometry using consistent settings for Cy3 fluorescence.
- Use the median fluorescence intensity (MFI) from each aliquot to calculate the plasmid count in each compartment [20]:
- Pni = MFI(A) - MFI(B)
- Pne = MFI(B) - MFI(C) (Note: This requires validation of the permeabilization method)
- Pcyto = MFI(B) - Pne (Conceptual)
- Convert fluorescence values to absolute plasmid counts per cell using a standard curve or qPCR validation.
- Fit the time-course data for Pcyto, Pne, and Pni to a four-compartment mass-action model using appropriate software to extract the rate constants kcell, kne, and kni [20].
4. Notes:
- Account for the pH-sensitivity of Cy3 fluorescence, as the signal can vary between the neutral cytosol and acidic endosomes [20].
- Validate the flow cytometry-based plasmid counts against a gold standard method like qPCR for initial setup [20].
- The rate constants obtained are specific to the vector, cell type, and experimental conditions.
Protocol: Tuning PEI/DNA Nanoparticles for Enhanced T-cell Transfection
This protocol outlines a systematic approach to optimize polyethyleneimine (PEI)/DNA nanoparticles for efficient gene delivery into hard-to-transfect human T cells [22].
1. Principle: By methodically adjusting the physicochemical properties of PEI/DNA polyplexes (N/P ratio), cell culture conditions, and the transfection protocol itself, gene delivery efficiency can be significantly enhanced.
2. Reagents and Equipment:
- Plasmid DNA
- Polyethylenimine (PEI), linear or branched.
- Human T cells.
- Cell culture media (e.g., RPMI 1640), transfection media.
- Dynamic Light Scattering (DLS) and Zeta Potential Analyzer.
3. Procedure: Step 1: Nanoparticle Formation and Characterization
- Form PEI/DNA polyplexes at various N/P ratios (e.g., from 5 to 10) in an opti-MEM or serum-free buffer.
- Incubate for 15-30 minutes at room temperature to allow complex formation.
- Characterize the formed nanoparticles using DLS for hydrodynamic size and zeta potential analyzer for surface charge. An N/P ratio of 8.0 is often a good starting point for optimization, yielding particles with a stable size and positive zeta potential [22].
Step 2: Optimization of Culture and Transfection Conditions
- Cell Seeding Density: Seed T cells at a high density (e.g., 1-2 x 10^6 cells/mL) at the time of transfection [22].
- Transfection Method: Employ a "reverse transfection in vials" method. Mix the polyplexes with a concentrated cell pellet in a small volume, incubate for a short period (e.g., 20-30 minutes), then add the mixture to complete culture media in the well. This method can dramatically increase uptake compared to direct addition of polyplexes to cells in culture plates [22].
- Post-Transfection Media Change: Add complete media shortly after (e.g., within 1-2 hours) transfection to support cell viability [22].
Step 3: Modulation of Cellular Physiology (Advanced)
- To further enhance transfection, resuspend the T cell pellet in a hypotonic extracellular media adjusted to pH 9.0 immediately before the reverse transfection step. This temporarily alters cell membrane permeability. Maintain cells in this medium only during the brief transfection incubation, then dilute with complete culture medium. This approach can dramatically boost transfection rates but requires careful optimization to minimize cytotoxicity [22].
4. Notes:
- Gel retardation assays should be performed to confirm complete DNA complexation at the chosen N/P ratio [22].
- The optimal DNA dosage and complex volume should be determined empirically.
- Always include controls for cytotoxicity (e.g., using MTS or LDH assays) when optimizing protocols, especially when using perturbative conditions like hypotonic/high pH treatment.
The Scientist's Toolkit: Key Research Reagents
Table 2: Essential Reagents for Non-Viral Gene Delivery Research
| Reagent / Material | Function / Role | Key Characteristics & Notes |
|---|---|---|
| Ionizable Cationic Lipids | Core component of LNPs; complexes nucleic acids, enables endosomal escape. | Positive charge at low pH for complexation, neutral at physiological pH for reduced toxicity. e.g., DLin-MC3-DMA. |
| Poly(beta-amino ester) (PBAE) | Biodegradable cationic polymer for DNA complexation. | High transfection efficiency in some cell types; tunable structure via synthesis. "Proton sponge" effect. |
| Polyethylenimine (PEI) | Cationic polymer for nucleic acid complexation. | High buffering capacity aids endosomal escape. Branched and linear forms vary in efficacy/toxicity. |
| Polyethylene Glycol (PEG) | Stealth agent; improves stability and pharmacokinetics. | Conjugated to lipids or polymers to reduce protein adsorption and extend circulation half-life. |
| Nuclear Localization Signal (NLS) | Peptide sequence that enhances nuclear import. | Covalently linked to vector or plasmid; binds to importin proteins. e.g., SV40 T-antigen NLS (PKKKRKV). |
| Cell-Penetrating Peptides (CPP) | Peptide sequences that enhance cellular uptake. | e.g., TAT peptide (GRKKRRQRRRPQ). Can be used to functionalize nanoparticles. |
| Targeting Ligands | Enables cell-specific targeting. | Includes folic acid, transferrin, antibodies, or aptamers. Conjugated to nanoparticle surface. |
| Cy3-dUTP or Label-IT Kits | Fluorescent labeling of nucleic acids for tracking. | Allows quantification of cellular uptake and intracellular trafficking via flow cytometry or microscopy. |
Enhancing the journey of non-viral gene delivery vectors from the cell surface to the nucleus requires a multi-faceted strategy grounded in quantitative data. Key approaches include engineering nanoparticles with optimized size (60-100 nm) and surface charge, incorporating functional components like PEG for stability and ionizable lipids/polymers for endosomal escape, and utilizing biological motifs such as NLS for nuclear import. The protocols provided for quantifying uptake rates and optimizing transfection in difficult cells offer a practical starting point for researchers. By systematically addressing each barrier with these detailed strategies, the development of more efficient and clinically viable non-viral gene therapies can be significantly accelerated.
Addressing Stability, Pharmacokinetics, and Repeat Dosing Potential
The advancement of gene delivery using non-viral nanoparticle vectors represents a paradigm shift in therapeutic strategies for genetic disorders, cancer, and a host of other diseases. Unlike viral vectors, non-viral systems, primarily based on lipid and polymer nanoparticles, offer the potential for enhanced safety profiles, reduced immunogenicity, and greater design flexibility [40]. However, their clinical translation and commercial viability are contingent upon overcoming three interconnected core challenges: maintaining product stability, understanding and optimizing in vivo pharmacokinetics (PK), and unlocking the potential for safe and effective repeat dosing. This application note details critical protocols and analytical frameworks to address these challenges, providing researchers with methodologies to characterize and improve these essential parameters within a comprehensive gene delivery development program.
Stability Challenges and Assessment Protocols
Stability is a critical quality attribute for non-viral gene therapies, encompassing both the physical integrity of the nanoparticle and the chemical stability of the encapsulated genetic payload. Key instability issues include particle aggregation, lipid or polymer degradation, and nuclease-mediated degradation of nucleic acids, all of which can severely compromise therapeutic efficacy [73].
Table 1: Key Stability Challenges for Non-Viral Nanoparticle Vectors
| Stability Challenge | Root Cause | Impact on Product | Common Stabilization Strategies |
|---|---|---|---|
| Physical Degradation | Particle aggregation, fusion, or precipitation during storage or shipping [73]. | Altered biodistribution, reduced cellular uptake, and potential safety issues. | Optimized buffer composition (e.g., sucrose, trehalose as cryoprotectants), controlled particle size distribution, lyophilization [73]. |
| Chemical Instability | Hydrolysis or oxidation of lipid/polymer components; degradation of PEG-lipid conjugates [73]. | Reduced encapsulation efficiency, inefficient endosomal escape, and loss of transfection potency. | Use of antioxidants, control of storage pH and temperature, formulation under inert atmosphere [73]. |
| Nucleic Acid Degradation | Ribonuclease (RNase) activity for mRNA; Deoxyribonuclease (DNase) activity for DNA [40] [73]. | Loss of therapeutic protein expression, abrogation of gene editing or silencing activity. | Complete encapsulation within nanoparticle, use of nuclease inhibitors in formulations, nucleotide modification (e.g., pseudouridine for mRNA) [40] [73]. |
Protocol: Comprehensive Stability Profiling of LNPs
This protocol outlines a methodology for assessing the critical physical and chemical stability parameters of lipid nanoparticle (LNP) formulations encapsulating mRNA or DNA.
I. Materials and Reagents
- LNP formulation for testing
- HPLC-grade water and buffers (e.g., PBS, Tris-EDTA)
- SYBR Gold or similar nucleic acid staining dye
- Ribonuclease A (for mRNA-LNP integrity assay)
- Dynamic Light Scattering (DLS) / Nanoparticle Tracking Analysis (NTA) instrument
- HPLC system with appropriate columns (e.g., C18 for lipid analysis)
II. Experimental Workflow
- Accelerated Stability Studies:
- Aliquot LNP formulations and store at recommended (e.g., 4°C, -80°C) and stressed conditions (e.g., 25°C, 40°C).
- Sample at predetermined time points (e.g., 0, 1, 2, 4 weeks) for analysis.
Physical Stability Assessment:
- Particle Size and PDI: Dilute LNPs in a suitable buffer and measure hydrodynamic diameter and polydispersity index (PDI) via DLS. A significant increase (>20%) indicates aggregation.
- Zeta Potential: Measure surface charge to monitor changes in colloidal stability.
- Encapsulation Efficiency: Use a fluorescent dye exclusion assay. Mix LNPs with dye, measure total fluorescence, then add a detergent to disrupt particles and measure fluorescence again. Encapsulation % = (1 - (Fpre-disruption / Fpost-disruption)) * 100.
Chemical Stability Assessment:
- Lipid/Polymer Integrity: Use Reverse-Phase HPLC to quantify the degradation of lipid components (e.g., ionizable lipid, PEG-lipid) against fresh standards.
- Nucleic Acid Integrity: For mRNA-LNPs, extract RNA and analyze via capillary electrophoresis (e.g., Fragment Analyzer) to quantify the percentage of full-length transcript.
The following workflow diagram illustrates the key steps in this stability assessment protocol:
Pharmacokinetic Modeling and Quantitative Analysis
The pharmacokinetic (PK) profile of non-viral nanoparticles is complex, involving administration, distribution to target tissues, cellular uptake, endosomal escape, and eventual expression or action of the genetic payload [74]. Model-Informed Drug Development (MIDD) approaches, including Quantitative Systems Pharmacology (QSP) and Physiologically Based Pharmacokinetic (PBPK) modeling, are increasingly vital for translating preclinical data into clinical predictions and optimizing dosing regimens [75].
Table 2: Key ADME Parameters for LNP-delivered mRNA Modalities
| PK Process | Key Characteristics | Influencing Factors |
|---|---|---|
| Absorption | After IM/SC injection, LNPs <200 nm drain to lymphatics before systemic circulation [74]. | Injection route, particle size, surface PEGylation, LNP composition. |
| Distribution | Widespread distribution to organs with high blood flow (liver, spleen, lungs); influenced by a formed "protein corona" [74]. | Surface charge, lipid composition, PEG density, target tissue endothelial permeability. |
| Metabolism & Elimination | mRNA degraded by ribonucleases; LNPs cleared by the mononuclear phagocyte system (MPS) in liver and spleen [74]. | Nuclease activity in tissue and blood, LNP stability, opsonization by serum proteins, immune recognition. |
Protocol: Developing a PBPK Model for LNP-mRNA Therapeutics
This protocol provides a framework for building a mechanistic PBPK model to simulate the in vivo journey of an LNP-mRNA therapeutic, from administration to protein expression.
I. Model Structure and Parameters The model structure should incorporate key physiological compartments (e.g., plasma, liver, spleen, muscle, lymph nodes) connected by blood flow. Critical system-specific parameters must be defined:
- LNP-related Parameters: Hydrodynamic diameter, surface charge (zeta potential), lipid composition, PEG-lipid percentage, encapsulation efficiency.
- mRNA-related Parameters: Nucleotide sequence, modification status, translation rate constant, degradation rate constant in cytoplasm.
- Physiological Parameters: Organ volumes and blood flow rates (allometric scaling from preclinical species to human), endosomal escape efficiency, immune activation constants.
II. Implementation and Workflow
- Model Formulation: Define the system of ordinary differential equations (ODEs) describing mass transfer between compartments, LNP cellular uptake, endosomal release, and mRNA translation and degradation.
- Parameter Estimation: Use preclinical PK/PD data (e.g., from mice and non-human primates) to estimate unknown parameters. Fit the model to time-course data of LNP concentration in plasma, mRNA levels in target tissues, and expressed protein levels in plasma or tissue.
- Model Validation and Simulation: Validate the model by comparing its predictions to a separate set of experimental data not used for parameter estimation. Once validated, perform simulations to predict first-in-human dosing, explore different dosing regimens, and identify critical parameters controlling exposure and efficacy.
The following diagram illustrates the core structure and processes of a PBPK model for LNP-mRNA therapeutics:
Strategies for Enabling Repeat Dosing
A significant limitation of many gene therapy vectors, including some non-viral systems, is the induction of anti-drug antibodies (ADAs) and anti-polymer antibodies (APAs) that can clear subsequent doses, diminishing efficacy and potentially causing adverse events [40] [76]. Enabling repeat dosing is therefore essential for chronic disease management and dose titration.
Protocol: Assessing and Mitigating Immunogenicity for Repeat Dosing
I. Materials and Reagents
- Test LNP formulation and control (e.g., empty LNPs)
- Relevant animal model (e.g., C57BL/6 mice, Sprague-Dawley rats)
- ELISA kits for detecting anti-PEG and anti-vector antibodies
- Flow cytometry reagents for immune cell profiling
- Adjuvants (if needed for immunogenicity studies)
II. Experimental Workflow
- Immunogenicity Study Design:
- Group animals (n≥5) to receive initial LNP dose via the intended clinical route.
- Collect serum samples pre-dose and at regular intervals post-dose (e.g., days 7, 14, 28).
- Administer a second, identical dose at a predetermined interval (e.g., 4 weeks).
- Monitor for signs of accelerated blood clearance (ABC) phenomenon upon second dose, evidenced by rapidly declining PK profiles.
Humoral Immune Response Assessment:
- Use direct or sandwich ELISA on collected serum to quantify titers of anti-PEG IgM and IgG antibodies.
- Develop an assay to detect antibodies against the core lipid or polymer components, if applicable.
Strategies to Mitigate Immunogenicity:
- Vector Stealthing: Employ low-immuneogenic lipids and polymers. Use alternative PEG-lipids (e.g., with different PEG chain lengths or architectures) or non-PEG surfactants [40].
- Dosing Interval Optimization: Data from the immunogenicity study can inform the minimal safe interval between doses to allow antibody titers to wane.
- Immunosuppressive Regimens: Co-administer transient immunosuppressants (e.g., dexamethasone) with the LNP dose to blunt the adaptive immune response [76].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Non-Viral Vector Research
| Reagent / Material | Function in Research | Example Application |
|---|---|---|
| Cationic Lipids | Electrostatic complexation with nucleic acids; forms core structure of LNPs; promotes endosomal escape via disruption [40] [74]. | SM-102, DLin-MC3-DMA used in commercial LNP formulations for mRNA delivery. |
| Ionizable Lipids | Positively charged at low pH (aiding encapsulation and endosomal escape) but neutral at physiological pH (reducing toxicity) [74]. | Critical component of modern LNPs for efficient in vivo mRNA delivery. |
| PEG-Lipids | Steric stabilization of nanoparticles; reduces aggregation and opsonization; modulates pharmacokinetics and biodistribution [74]. | DMG-PEG2000, DSG-PEG2000 used to control LNP size and circulation time. |
| Cationic Polymers | Condense nucleic acids into polyplexes; can enhance cellular uptake and provide buffering capacity for endosomal escape [40] [77]. | Polyethyleneimine (PEI), Poly(lactic-co-glycolic acid) (PLGA), Chitosan. |
| Stabilizing Excipients | Protect nanoparticle integrity and nucleic acid payload during storage and freeze-thaw cycles [73]. | Sucrose, trehalose, mannitol used as cryoprotectants and lyoprotectants. |
The field of gene delivery is increasingly leveraging non-viral nanoparticle vectors to overcome the inherent limitations of viral vectors, such as immunogenicity, insertional mutagenesis, and limited cargo capacity [62] [17]. Advanced formulation design has evolved to create sophisticated hybrid and smart nanoparticle systems that integrate multiple material classes and exhibit responsive behaviors. These systems are engineered to navigate the complex biological barriers to gene delivery, from extracellular hurdles like enzymatic degradation and opsonization to intracellular challenges including endosomal escape and nuclear entry [62]. By rationally combining lipids, polymers, and inorganic materials, hybrid nanoparticles achieve superior performance characteristics unattainable by single-component systems, while smart nanoparticles respond to specific physiological stimuli for spatiotemporally controlled gene release [78] [79]. This document provides detailed application notes and experimental protocols for the design, fabrication, and characterization of these advanced systems within the context of non-viral gene delivery research.
Core Components and Quantitative Properties of Nanoparticle Systems
The rational design of nanoparticle systems begins with the selection of core components whose physicochemical properties dictate biological interactions and therapeutic efficacy. The size, surface charge, and composition of nanoparticles are primary determinants of their stability, cellular uptake efficiency, and intracellular trafficking [62] [80].
Table 1: Key Characteristics of Major Nanoparticle Platforms for Gene Delivery
| Platform Type | Common Materials | Typical Size Range | Surface Charge (Zeta Potential) | Key Advantages | Primary Limitations |
|---|---|---|---|---|---|
| Lipid-Based | LNPs (ionizable lipids, phospholipids, cholesterol, PEG-lipids), Liposomes | 50-200 nm [80] | Slightly negative to mildly positive | High encapsulation efficiency for nucleic acids; Proven clinical success (mRNA vaccines) [81] [62] | Variable stability; Potential immunogenicity with PEG [82] |
| Polymer-Based | PEI, PAMAM dendrimers, PLGA, Chitosan | 50-300 nm [17] | Highly positive for polycations (e.g., PEI) | High nucleic acid condensation; "Proton sponge" endosomal escape [62] [17] | Cytotoxicity at high charge densities; Batch-to-batch variability [82] |
| Inorganic | Gold nanospheres/rods, Iron oxide, Silica | 10-150 nm | Variable (often functionalized) | Tunable size/shape; Unique optical/magnetic properties | Potential long-term toxicity; Biopersistence concerns [47] |
| Hybrid | Polymer-Lipid, Polymer-Inorganic, Lipid-Inorganic | 70-250 nm | Tailorable | Combines advantages of components; Enhanced functionality | More complex manufacturing; Additional characterization requirements [79] |
Nanoparticle size plays a particularly critical role in regulating biodistribution, cellular uptake, and transport mechanisms [80]. Data-driven optimization approaches, such as the Prediction Reliability Enhancing Parameter (PREP), have demonstrated that target sizes of approximately 100 nm for compressible microgels and 170 nm for polyelectrolyte complexes are optimal for biological penetration and circulatory stability, respectively [80]. Surface charge, measured as zeta potential, influences not only cellular uptake but also serum stability, with highly positive charges often leading to nonspecific protein adsorption and rapid clearance [62].
Table 2: Impact of Nanoparticle Physicochemical Properties on Biological Behavior
| Physicochemical Property | Optimal Range for Gene Delivery | Biological Consequences | Measurement Techniques |
|---|---|---|---|
| Hydrodynamic Size | 50-200 nm [62] [80] | <50 nm: Rapid renal clearance; 50-150 nm: Enhanced tissue penetration; >200 nm: Increased immune recognition and sequestration | Dynamic Light Scattering (DLS) |
| Zeta Potential | Slightly negative to mildly positive (+5 to -20 mV) [62] | Highly positive: Enhanced cell uptake but cytotoxicity; Highly negative: Reduced uptake but improved stability | Laser Doppler Velocimetry |
| Polydispersity Index (PDI) | <0.2 [80] | Low PDI indicates uniform size distribution, predictable behavior | Dynamic Light Scattering (DLS) |
| Surface Functionalization | Targeting ligands (e.g., transferrin, RGD peptides) | Enhanced cell-specific uptake; Receptor-mediated transcytosis [83] [17] | HPLC, Mass Spectrometry, ELISA |
Advanced Hybrid System Design and Fabrication Protocols
Layer-by-Layer (LbL) Polyelectrolyte Nanoparticles
Principle: This technique involves the sequential deposition of oppositely charged polyelectrolytes onto a nanoparticle core, creating a multilayered shell that enables precise control over gene release kinetics and enhances stability [79].
Protocol:
- Core Nanoparticle Preparation: Begin with 100 nm cationic poly(D,L-lactide) (PLA) core nanoparticles at a concentration of 5 mg/mL in 10 mM HEPES buffer (pH 7.4).
- First Layer Deposition: Add an equal volume of 1 mg/mL heparin (anionic polyelectrolyte) solution containing 0.5 M NaCl to the core nanoparticle suspension. The salt concentration is critical for controlling layer thickness and structure [79]. Incubate with gentle rotation for 20 minutes at room temperature.
- Washing Step: Remove excess polyelectrolyte by centrifugation at 15,000 × g for 15 minutes. Resuspend the pellet in 10 mM HEPES buffer.
- Second Layer Deposition: Add an equal volume of 1 mg/mL polyethyleneimine (PEI, cationic polyelectrolyte) solution to the suspension. Incubate with gentle rotation for 20 minutes.
- Repeat Washing: Centrifuge at 15,000 × g for 15 minutes and resuspend in buffer.
- Additional Layers: Repeat steps 2-5 until the desired number of layers is achieved (typically 3-5 layers).
- Gene Loading: Incubate the final LbL nanoparticles with plasmid DNA or mRNA at a weight ratio of 10:1 (nanoparticle:gene) for 30 minutes to form complexes.
Quality Control: Monitor the increase in hydrodynamic diameter (≈10-15 nm per layer) and charge reversal (from positive to negative with each layer) using dynamic light scattering after each deposition step [79].
Lipid-Polymer Hybrid Nanoparticles (LPNs)
Principle: LPNs combine the structural integrity and controlled release properties of polymeric cores with the biomimetic properties and fusogenic capabilities of lipid shells, enhancing gene delivery efficiency [78].
Protocol:
- Polymer Core Formation: Prepare PLGA nanoparticles using a nanoemulsion method. Dissolve 100 mg PLGA in 5 mL ethyl acetate. Add this organic phase to 20 mL of 2% polyvinyl alcohol (PVA) aqueous solution and emulsify using a probe sonicator (100 W, 2 minutes on ice).
- Solvent Evaporation: Stir the emulsion overnight at room temperature to evaporate the organic solvent. Collect the PLGA nanoparticles by centrifugation at 20,000 × g for 20 minutes and wash twice with distilled water.
- Lipid Shell Formation: Prepare a lipid film by evaporating a chloroform solution containing lecithin, cholesterol, and PEG-lipid (60:35:5 molar ratio) under reduced pressure using a rotary evaporator.
- Hydration and Fusion: Hydrate the lipid film with the PLGA nanoparticle suspension (5 mg/mL) and subject to 5 cycles of freeze-thaw (liquid nitrogen to 40°C water bath) to promote lipid fusion around the polymeric core.
- Size Reduction: Extrude the suspension through polycarbonate membranes (400 nm, then 200 nm) using a mini-extruder to achieve uniform size distribution.
- Gene Encapsulation: For gene loading, dissolve the nucleic acid in the aqueous phase during the initial emulsion step (for DNA) or employ a post-loading adsorption method (for siRNA/mRNA) by incubating the formed LPNs with genes at optimal weight ratios.
Quality Control: Determine encapsulation efficiency using a Quant-iT PicoGreen assay for unencapsulated DNA in the supernatant after centrifugation. Effective LPNs should achieve >85% encapsulation efficiency.
Signaling Pathways and Experimental Workflows
The following diagram illustrates the systematic design approach for creating hybrid and smart nanoparticle systems, from material selection through biological evaluation:
Diagram 1: Systematic design workflow for hybrid nanoparticle systems, illustrating the iterative process integrating the Four-Domain Model for rational design [78] [80].
The cellular internalization and intracellular trafficking of hybrid nanoparticles involves multiple pathways and barriers as shown below:
Diagram 2: Cellular uptake mechanisms and intracellular trafficking pathways for gene-loaded nanoparticles, highlighting key barriers including endosomal entrapment and nuclear membrane penetration [62] [17].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Hybrid Nanoparticle Development
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Cationic Polymers | Branched PEI (25 kDa), PAMAM Dendrimers (G5-G7) | Nucleic acid condensation; Proton sponge endosomal escape [17] | Cytotoxicity increases with molecular weight and charge density; requires biodegradability modifications |
| Ionizable Lipids | DLin-MC3-DMA, SM-102, ALC-0315 | pH-dependent charge for siRNA/mRNA encapsulation and endosomal release [62] | Optimal pKa ~6.4; critical for in vivo efficacy; component of FDA-approved LNPs |
| Biodegradable Polymers | PLGA (50:50 to 85:15 LA:GA), Chitosan | Core matrix for controlled release; improved biocompatibility [82] [17] | LA:GA ratio affects degradation rate; molecular weight influences drug release kinetics |
| Surface Stabilizers | PEG-lipids (DMG-PEG2000, ALC-0159), Poloxamers | Steric stabilization; reduces protein adsorption and extends circulation [82] | PEG density affects pharmacokinetics; potential for anti-PEG antibodies with repeated dosing |
| Targeting Ligands | Transferrin, RGD peptides, Folate, Antibodies | Active targeting to specific cell types or tissues [83] [17] | Ligand density optimization critical; avoid steric hindrance from PEG corona |
| Stimuli-Responsive Materials | pH-sensitive (acetal derivatives), Redox-sensitive (disulfide bonds) | Triggered drug release in specific microenvironments (e.g., tumor, endosome) [78] | Must demonstrate clear advantage over passive release; sensitivity must match pathophysiology |
| Characterization Tools | Dynamic Light Scattering, HPLC with CAD/ELSD, PicoGreen Assay | Size, charge, and encapsulation efficiency quantification [80] [84] | Multi-method approach recommended; consider both number and intensity distributions for size |
Data-Driven Optimization and Characterization Protocols
PREP (Prediction Reliability Enhancing Parameter) Optimization
Principle: PREP is a data-driven modeling approach that combines multiple model alignment metrics to enhance predictive reliability in nanoparticle design, significantly reducing experimental iterations needed to achieve target properties [80].
Protocol:
- Initial Dataset Establishment: Compile historical synthesis data including at least 3 input variables (e.g., monomer concentration, crosslinker density, surfactant concentration) and corresponding output variables (hydrodynamic size, PDI, zeta potential). A minimum of 10-15 data points is recommended.
- Latent Variable Model (LVM) Development:
- Preprocess data using mean-centering and variance scaling.
- Perform Partial Least Squares (PLS) regression to identify latent variables connecting input parameters to output properties.
- Validate model using leave-one-out cross-validation.
- Target Definition: Specify desired nanoparticle properties (e.g., size = 100 nm, PDI < 0.2, zeta potential = -15 mV).
- PREP Application:
- Apply PREP metric to identify optimal synthesis parameters within the model's latent space.
- Calculate PREP using the formula that combines Hotelling's T² and Squared Prediction Error (SPE) statistics.
- Select input parameters that minimize PREP value while achieving target outputs.
- Experimental Validation: Synthesize nanoparticles using PREP-predicted parameters.
- Iterative Refinement: Compare experimental results with predictions and update the dataset and model accordingly. Typically, 2-3 iterations are sufficient to achieve target properties [80].
Advanced Characterization Techniques
Quantifying Gene Delivery Efficiency:
- Cellular Uptake Measurement:
- Label nanoparticles with Cy5 fluorophore (0.5% w/w).
- Incubate with cells (50,000 cells/well in 24-well plates) for 4 hours at 37°C.
- Wash with cold PBS, trypsinize, and analyze by flow cytometry.
- Include inhibitors (e.g., chlorpromazine for clathrin-mediated endocytosis, methyl-β-cyclodextrin for caveolae-mediated endocytosis) to determine uptake pathways [62].
- Endosomal Escape Quantification:
- Transfect cells with Gal8-mCherry reporter construct.
- Treat with nanoparticles and image using confocal microscopy.
- Quantify Gal8-positive puncta as indicator of endosomal damage/escape.
- Gene Expression Analysis:
- For mRNA delivery: Measure protein expression by flow cytometry or Western blot 24 hours post-transfection.
- For plasmid DNA: Quantify reporter gene expression 48-72 hours post-transfection.
- Normalize to total protein content and report as mean fluorescence intensity or luminescence units per mg protein.
The rational design of hybrid and smart nanoparticle systems represents a paradigm shift in non-viral gene delivery, moving from single-component formulations to sophisticated, multi-functional architectures. The integration of material sciences, data-driven optimization, and biological insights enables researchers to engineer nanoparticles with enhanced efficacy and specificity. The protocols and application notes provided herein establish a framework for developing these advanced systems, with emphasis on reproducible fabrication methods, comprehensive characterization, and functional validation. As the field progresses, the convergence of these approaches with personalized medicine and artificial intelligence will further accelerate the development of next-generation gene delivery platforms for clinical application.
Non-Viral vs. Viral Vectors: A Critical Analysis of Safety, Efficacy, and Clinical Utility
Within the framework of advancing non-viral nanoparticle vectors for gene delivery, a critical assessment of safety profiles is paramount. This application note provides a detailed, side-by-side comparison of two primary safety concerns in gene therapy: immunogenicity (the potential to provoke an immune response) and insertional mutagenesis (the risk of unintended genomic insertions that disrupt gene function). While viral vectors are the established workhorse of clinical gene therapy, their non-viral counterparts offer distinct safety advantages that are crucial for long-term therapeutic success. This document summarizes quantitative data in structured tables, outlines definitive experimental protocols for risk assessment, and provides visualization tools to aid researchers and drug development professionals in the selection and de-risking of gene delivery platforms.
Comparative Analysis: Immunogenicity
Immunogenicity remains a significant barrier to effective gene therapy, influencing both patient safety and treatment efficacy. The innate and adaptive immune responses elicited by viral and non-viral vectors differ substantially in their mechanisms and consequences.
Table 1: Head-to-Head Comparison of Immunogenicity Profiles
| Vector Type | Key Immune Activators | Primary Immune Response | Clinical Consequences | Mitigation Strategies |
|---|---|---|---|---|
| Adeno-Associated Virus (AAV) | Capsid proteins; Transgene product [85] [1] | Adaptive Immunity: Neutralizing antibody formation; T-cell-mediated clearance of transduced cells [85] [1] | Limited re-administration efficacy; Potential hepatotoxicity [1] | Capsid engineering; Transient immunosuppression; Tissue-specific promoters [1] |
| Adenovirus (Ad) | Capsid proteins; Viral DNA [85] | Strong Innate & Adaptive Immunity: Robust inflammation; high-level antibody production [85] | Acute inflammatory toxicity (e.g., fever, hypotension); High immunogenicity limits use [85] [1] | Confined to applications where immunity is beneficial (e.g., vaccines, oncolytics) [1] |
| Lentivirus (LV) | Viral RNA [85] | Innate Immunity: Recognized by intracellular PRRs [85] | Lower immunogenicity than Ad; concerns in ex vivo settings [86] [85] | Use of non-human lentiviruses (e.g., SIV) to reduce recognition [85] |
| Lipid Nanoparticles (LNPs) | Ionizable lipids; PEG-lipids [87] | Innate Immunity: Inflammasome activation (IL-1β, IL-6); Type I Interferon response; CARPA [87] | Injection-site reactions, fever, fatigue; Rare anaphylaxis [87] | Optimizing lipid structure; Adjusting PEG-lipid content; Pre-medication [87] |
| Cationic Polymers (e.g., PEI) | Cationic charge [62] [85] | Innate Immunity: Inflammatory cytokine release; Complement activation [62] | Concentration-dependent cytotoxicity [62] [85] | Polymer modification (e.g., PEGylation); Use of biodegradable variants [62] [85] |
Key Experimental Protocol: Assessing LNP-Induced Innate Immunity
Objective: To evaluate the potential of Lipid Nanoparticles (LNPs) to activate innate immune pathways in vitro.
Materials:
- Test Articles: Empty LNPs (without nucleic acid cargo) and RNA-loaded LNPs.
- Control Articles: Phosphate-Buffered Saline (PBS) and a known immunostimulant (e.g., LPS).
- Cell Line: Human peripheral blood mononuclear cells (PBMCs) or a macrophage cell line (e.g., THP-1-derived macrophages).
- Key Reagents: ELISA kits for human IL-6, TNF-α, and IFN-β; cell culture media and supplements.
Procedure:
- Cell Seeding and Differentiation: Seed PBMCs or differentiate THP-1 cells into macrophages in 24-well plates.
- Treatment: Treat cells with a range of concentrations of empty LNPs, RNA-loaded LNPs, PBS (negative control), and LPS (positive control). Incubate for 6-24 hours.
- Supernatant Collection: Collect cell culture supernatant by centrifugation.
- Cytokine Analysis: Use commercially available ELISA kits according to the manufacturer's instructions to quantify the levels of IL-6, TNF-α, and IFN-β in the supernatant.
- Data Interpretation: Compare cytokine levels from LNP-treated groups to controls. A statistically significant increase in cytokines indicates innate immune activation.
Signaling Pathway Visualization: LNP Immunogenicity
The following diagram illustrates the primary innate immune signaling pathways activated by Lipid Nanoparticles, integrating key data from the comparative analysis.
Diagram 1: LNP-Induced Innate Immune Signaling. Lipid Nanoparticles (LNP) can be recognized by Toll-like Receptors (TLR7/8) in the endosome, leading to NF-κB activation and inflammasome formation, and by RIG-I/MDA5 in the cytosol, leading to IRF3-mediated Type I Interferon production. These pathways converge to establish a pro-inflammatory state [87].
Comparative Analysis: Insertional Mutagenesis
Insertional mutagenesis refers to the disruption of host gene function or regulation caused by the integration of a therapeutic vector into the genome. This risk is a critical differentiator between vector platforms.
Table 2: Head-to-Head Comparison of Insertional Mutagenesis Risks
| Vector Type | Integration Mechanism | Genomic Integration Profile | Reported Clinical Risks | Risk Mitigation Strategies |
|---|---|---|---|---|
| Retrovirus (RV) | Viral integrase; random integration [85] | Preferential integration near transcriptional start sites [85] | Cases of leukemogenesis in early SCID trials [85] | Self-inactivating (SIN) designs with deleted enhancer/promoter elements in LTRs [85] |
| Lentivirus (LV) | Viral integrase; semi-random integration [86] [85] | Prefers integration into active transcriptional units [85] | Myelodysplastic syndrome reported post-Skysona therapy [1] | Use of SIN designs; safer modern generations derived from non-human viruses [85] |
| Adeno-Associated Virus (AAV) | Predominantly non-integrating (episomal) [85] | Rare, non-targeted integration via non-homologous end joining [85] | Theoretical risk; no significant clinical reports to date [85] [1] | Natural preference for episomal persistence minimizes risk. |
| Non-Viral Vectors (LNPs, Polymers) | Non-integrating by design [8] [62] [1] | No genomic integration intended. | Negligible risk of insertional mutagenesis [8] [62] [1] | Transient expression is a inherent safety feature; no specific mitigation required. |
Key Experimental Protocol: Assessing Genomic Integration Sites
Objective: To map the genomic integration sites of viral vectors in vitro to assess potential genotoxicity.
Materials:
- Test Article: Cells transduced with the lentiviral or retroviral vector of interest.
- Control Article: Untreated cells.
- Key Kits/Reagents: Genomic DNA extraction kit; LAM-PCR or Lenti-X Integration Site Analysis Kit (Takara Bio); Next-Generation Sequencing (NGS) library preparation kit; Bioinformatics software (e.g., R, Python with relevant packages).
Procedure:
- Cell Transduction and Culture: Transduce target cells (e.g., hematopoietic stem cells) at a low Multiplicity of Infection (MOI) to ensure single integration events. Culture cells for 2-3 weeks to allow for stable integration and expansion.
- Genomic DNA Extraction: Harvest cells and extract high-molecular-weight genomic DNA.
- Linear Amplification-Mediated PCR (LAM-PCR):
- Digestion: Use a restriction enzyme to digest genomic DNA.
- Linker Ligation: Ligate a known linker sequence to the digested ends.
- Nested PCR: Perform two rounds of PCR using primers specific to the linker and the viral Long Terminal Repeat (LTR) sequence.
- NGS Library Prep and Sequencing: Purify the PCR products, prepare an NGS library, and sequence on an appropriate platform.
- Bioinformatic Analysis: Map the sequenced reads to the human reference genome. Identify the genomic location of each integration site and analyze for enrichment in oncogenes or tumor suppressor genes.
Risk Assessment Visualization: Insertional Mutagenesis
The following workflow diagram outlines the key steps for evaluating the risk of insertional mutagenesis in a preclinical setting.
Diagram 2: Workflow for Integration Site Analysis. The process begins with transducing cells at a low multiplicity of infection (MOI) to isolate single integration events. After cell expansion, genomic DNA is analyzed via LAM-PCR and NGS to map integration sites and assess genotoxic risk based on proximity to transcriptional start sites (TSS) and oncogenes [85].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Vector Safety Assessment
| Reagent / Material | Function in Analysis | Specific Example(s) |
|---|---|---|
| ELISA Kits | Quantification of specific cytokines and chemokines in cell supernatant or serum to measure immune activation. | Human IL-6 ELISA Kit, Human IFN-β ELISA Kit [87] |
| Pattern Recognition Receptor (PRR) Reporter Cell Lines | To identify which specific innate immune pathway is activated by a vector. | HEK-Blue TLR7 cells, HEK-Blue TLR9 cells. |
| LAM-PCR Kit | Standardized method for the amplification and identification of viral vector integration sites from genomic DNA. | Lenti-X Integration Site Analysis Kit (Takara Bio) [85] |
| Cationic Polymer Transfection Reagents | "Gold standard" non-viral transfections for comparison; known for high efficiency but also cytotoxicity and immunogenicity. | Polyethylenimine (PEI), both linear and branched forms [62] [17] [85] |
| Lipid Nanoparticle Formulations | Pre-formulated LNPs for delivering RNA payloads (e.g., mRNA, siRNA) as a benchmark for non-viral performance and immunogenicity. | LNP systems containing ionizable lipids, phospholipids, cholesterol, and PEG-lipids [8] [87] |
| Next-Generation Sequencing (NGS) Platforms | Critical for high-throughput analysis of integration sites (genotoxicity) and transcriptomic changes (immunogenicity). | Illumina MiSeq/HiSeq for sequencing LAM-PCR products. |
The choice between viral and non-viral gene delivery vectors involves a direct trade-off between efficiency and safety. Viral vectors, particularly LV and RV, offer stable transduction but carry non-trivial risks of immunogenicity and insertional mutagenesis that require sophisticated mitigation and monitoring. In contrast, non-viral vectors, especially LNPs, present a markedly improved safety profile regarding genotoxicity, with immunogenicity that is primarily innate and transient, making it more manageable. As the field progresses, the combination of novel non-viral platforms with tissue-specific regulatory elements is a promising strategy to achieve targeted delivery without compromising safety, ultimately expanding the reach of gene therapies to a broader range of diseases.
Cargo Capacity and Versatility for Different Gene Therapy Modalities
The success of gene therapy is fundamentally constrained by the delivery vehicle's ability to transport diverse genetic payloads to target cells. Vectors require sufficient cargo capacity to accommodate therapeutic genes and regulatory elements, and versatility to deliver different modalities, from simple transgenes to complex gene-editing systems. The choice between viral and non-viral vectors represents a critical trade-off between delivery efficiency, payload size, and safety profile [14] [1].
Viral vectors, particularly Adeno-associated viruses (AAV), have been the workhorse for in vivo gene therapy but are limited to a cargo capacity of approximately 4.7 kilobases (kb) [1] [88]. This restriction excludes them from delivering large genes or complex multi-component systems. In contrast, emerging non-viral platforms like lipid nanoparticles (LNPs) offer substantially larger and more flexible cargo capacity, reported to encapsulate payloads of at least 10 kb [89], enabling new therapeutic modalities previously impossible with viral systems.
Comparative Analysis of Vector Cargo Capacities
Table 1: Cargo Capacity and Compatibility with Therapeutic Modalities for Different Vector Platforms
| Vector Platform | Therapeutic Cargo Types | Approximate Cargo Capacity | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Adeno-Associated Virus (AAV) | cDNA for transgene expression, shRNA | ≤ 4.7 kb [1] [88] | Established clinical safety profile; Efficient transduction of non-dividing cells | Limited cargo size restricts application for larger genes; Pre-existing immunity concerns |
| Lentivirus (LV) | cDNA, shRNA, CRISPR-Cas9 systems | ~8 kb [1] | Stable genomic integration enabling long-term expression; Suitable for ex vivo applications | Risk of insertional mutagenesis; Primarily used for ex vivo applications |
| Adenovirus (Ad) | cDNA, vaccines | ~8-36 kb [1] | Very high transduction efficiency; Large cargo capacity | Significant immunogenicity limits repeated administration |
| Lipid Nanoparticles (LNPs) | mRNA, siRNA, plasmid DNA, CRISPR-Cas9 components (mRNA + gRNA), proteins [90] [89] [88] | ≥ 10 kb [89] (with essentially unrestricted capacity) | Vast cargo flexibility; Low immunogenicity enabling redosing; Scalable manufacturing | Predominant liver tropism requires engineering for other targets; Transient expression profile |
Table 2: Quantitative Performance Metrics of DNA-LNPs versus Other Platforms
| Parameter | DNA-LNPs | mRNA-LNPs | AAV Vectors |
|---|---|---|---|
| Expression Duration | Months to years [90] | Days to weeks | Months to years (potentially lifelong) |
| Onset of Expression | Delayed (requires nuclear entry) | Rapid (cytosolic translation) | Moderately delayed |
| Risk of Insertional Mutagenesis | None [89] | None | Low but present [1] |
| Dosing Regimen | Amenable to redosing [89] [88] | Amenable to redosing | Limited by immune response |
| T Cell Response (Vaccine Context) | Superior CD8+ T cell responses relative to mRNA-LNPs [91] | Strong CD4+ T cell and antibody responses | Varies by serotype and administration route |
Overcoming the cGAS-STING Pathway in Plasmid DNA-LNP Delivery
A significant challenge in using plasmid DNA (pDNA)-LNPs is their propensity to activate the cGAS-STING pathway, an innate immune system mechanism that detects cytosolic DNA [90]. When standard pDNA-LNPs enter the cell cytoplasm, leaked pDNA can be recognized by cGAS (cyclic GMP-AMP synthase), which produces the second messenger cGAMP. This activates STING (Stimulator of Interferon Genes), leading to the production of type I interferons and pro-inflammatory cytokines such as IL-6, resulting in severe inflammatory responses [90]. In preclinical models, this activation has been linked to significant toxicity, including 100% mortality in mice at standard doses [90].
Strategic Inhibition for Safer pDNA-LNP Delivery
Inspired by mechanisms employed by DNA viruses to evade immune detection, researchers have developed a strategy to co-load pDNA-LNPs with nitro-oleic acid (NOA), an endogenous electrophilic lipid that covalently modifies STING and inhibits its activation [90]. These NOA-pDNA-LNPs effectively mitigate inflammatory responses in vitro and prevent mortality in vivo, without compromising long-term transgene expression, which can be sustained for several months [90].
Table 3: Key Research Reagent Solutions for pDNA-LNP Formulation and Characterization
| Reagent/Equipment | Function/Application | Specific Examples |
|---|---|---|
| Ionizable Lipid | Forms core structure of LNP; Enables endosomal escape | SM-102 [90] |
| Helper Lipid | Stabilizes LNP structure | DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine) [90] |
| PEG Lipid | Provides steric stabilization; Controls particle size | DMG-PEG 2000 [90] |
| Structural Lipid | Modulates membrane fluidity and integrity | Cholesterol [90] |
| Anti-inflammatory Lipid | Inhibits immune activation | Nitro-oleic acid (NOA) [90] |
| Microfluidic Instrument | Enables reproducible, scalable LNP production | NanoAssemblr Ignite [90] |
| Characterization Instrument | Measures particle size and polydispersity | Zetasizer Pro ZS (Malvern Panalytical) [90] |
The following diagram illustrates the immune activation pathway triggered by standard pDNA-LNPs and the strategic inhibition point of NOA:
Experimental Protocol: Production of Anti-inflammatory pDNA-LNPs
This protocol details the production of plasmid DNA-loaded lipid nanoparticles co-loaded with nitro-oleic acid (NOA) to enable safe, long-term gene expression by mitigating cGAS-STING pathway activation [90].
Materials and Reagents
- Lipids: SM-102 (ionizable lipid), Cholesterol, DSPC (helper lipid), DMG-PEG 2000 (PEG lipid), Nitro-oleic acid (NOA, anti-inflammatory lipid) [90]
- Genetic Material: Plasmid DNA (e.g., pALD-CV77-Luciferase) dissolved in nuclease-free water or TE buffer at 1 mg/mL [90]
- Solvents and Buffers: 200 proof ethanol, DEPC water, Phosphate-buffered saline (1× PBS, filter-sterilized), Citrate buffer (50 mM, pH 4) [90]
- Equipment: NanoAssemblr Ignite instrument (for microfluidics) or Vortex-Genie 2 (for vortex mixing), Zetasizer Pro ZS for particle size measurement, dialysis tubing (12-14 kD), 2 mL Eppendorf tubes, 15 mL conical tubes [90]
Procedure
Preparation of Lipid Mix
- Thaw and Prepare Lipid Stocks: Thaw all lipid components to room temperature. Dissolve lipids in 100% ethanol to the stock concentrations specified in Table 4.
- Create Lipid Mixture: In a 2 mL Eppendorf tube, combine the lipids and ethanol according to the volumes specified for the desired production scale (microfluidics for in vivo, vortex mixing for in vitro applications) as detailed in Table 4.
Table 4: Lipid Mix Formulation for NOA-pDNA-LNP Production (Adapted from [90])
| Component | Name | Stock Concentration (mg/mL) | Molar (%) in LNP | Volume (μL) for 0.4 mL LNPs (Microfluidics) | Volume (μL) for 0.16 mL LNPs (Vortex) |
|---|---|---|---|---|---|
| Ionizable Lipid | SM-102 | 50 | 50 | 35.5 | 4.4 |
| Cholesterol | Cholesterol | 11.6 | 38.5 | 64.2 | 8.0 |
| Helper Lipid | DSPC | 10 | 10 | 19.8 | 2.5 |
| PEG Lipid | DMG-PEG 2000 | 10 | 1.5 | 18.8 | 2.4 |
| Anti-inflammatory Lipid | NOA | 13.5 | 0.2 NOA:L (mol:mol) | 24.2 | 3.03 |
| Solvent | Ethanol | - | - | 37.5 | 79.69 |
Note: The "L" in NOA:L indicates the total lipid content excluding NOA. This protocol is designed for a 40:1 w/w ratio of total lipids to DNA (excluding NOA in the total lipid weight) [90].
Preparation of DNA Mix
- Prepare DNA Solution: Ensure plasmid DNA is at a concentration of 1 mg/mL in nuclease-free water or TE buffer.
- Create DNA Mixture: In a separate 2 mL Eppendorf tube, combine DNA and citrate buffer according to the volumes in Table 5.
Table 5: DNA Mix Formulation for NOA-pDNA-LNP Production (Adapted from [90])
| Component | Name | Composition | Volume (μL) for 0.4 mL LNPs (Microfluidics) | Volume (μL) for 0.16 mL LNPs (Vortex) |
|---|---|---|---|---|
| DNA | pALD-CV77-Luciferase | 1 mg/mL in water | 50.7 | 3.9 |
| Buffer | Citrate buffer | 50 mM, pH 4 | 348.3 | 116.1 |
LNP Production Using Microfluidics
- Setup: Insert a microfluidic cartridge into the NanoAssemblr Ignite instrument.
- Load Syringes: Draw the lipid mix (ethanol phase) into one 1 mL syringe and the DNA mix (aqueous phase) into another 1 mL syringe.
- Mixing: Connect both syringes to the cartridge and initiate the mixing process. The instrument forces the two phases through the microfluidic channels at a defined flow rate ratio (typically 3:1 aqueous:ethanol, vol%), resulting in the instantaneous formation of LNPs [90].
Downstream Processing and Characterization
- Dialysis: Transfer the formed LNP suspension to dialysis tubing (12-14 kD MWCO) and dialyze against 1× PBS, pH 7.4, for at least 2 hours at room temperature to remove residual ethanol and adjust the pH.
- Characterization:
- Particle Size and PDI: Use dynamic light scattering (e.g., Zetasizer Pro ZS) to measure the hydrodynamic diameter and polydispersity index (PDI). Quality LNPs should have a size of approximately 70 ± 5 nm with a PDI ≤ 0.2 [90].
- Encapsulation Efficiency: Use the Quant-iT PicoGreen dsDNA assay to quantify the percentage of plasmid DNA successfully encapsulated within the LNPs. Quality preparations should achieve >85% encapsulation efficiency [90].
The following workflow diagram summarizes the key steps in the LNP production process:
The evolution of gene therapy vectors reflects a continuous effort to balance cargo capacity, delivery efficiency, and safety. While viral vectors like AAV remain dominant in the current clinical landscape, their inherent cargo limitations restrict their application for larger genes and complex gene-editing tools. Non-viral vectors, particularly LNPs, offer a promising alternative with their superior cargo flexibility, capacity for multiple payload types, and improved safety profile enabling redosing. The development of sophisticated formulations such as NOA-pDNA-LNPs that strategically circumvent innate immune recognition demonstrates the field's progression toward overcoming significant biological barriers. As vector engineering continues to advance, the expanding toolbox of delivery platforms will undoubtedly unlock new therapeutic possibilities, ultimately enabling gene therapies for a broader spectrum of genetic diseases.
Manufacturing Complexity, Scalability, and Cost-Effectiveness
Application Note: Comparative Analysis of Non-Viral Nanoparticle Manufacturing
The transition from viral to non-viral nanoparticle vectors represents a paradigm shift in gene therapy manufacturing, addressing critical challenges in production complexity, scalability, and cost. This application note provides a structured comparison of major non-viral vector platforms and detailed protocols for their development and assessment.
Quantitative Comparison of Non-Viral Nanoparticle Platforms
Table 1: Performance Characteristics of Major Non-Viral Nanoparticle Platforms
| Platform | Manufacturing Complexity | Scalability Potential | Relative Cost | Key Advantages | Primary Limitations |
|---|---|---|---|---|---|
| Lipid-Based Nanoparticles (LNPs) | Moderate | High | Moderate | Rapid self-assembly, clinical validation for mRNA delivery, high encapsulation efficiency | Stability challenges, limited targeting capability, component variability [40] [9] |
| Polymer-Based Nanoparticles | Moderate to High | Moderate | Low to Moderate | Tunable release kinetics, chemical versatility, high nucleic acid loading | Potential cytotoxicity (e.g., PEI), batch-to-batch variability, polydisperse formulations [40] [92] |
| Inorganic Nanoparticles | Low to Moderate | High | Low | Excellent stability, precise size control, facile surface functionalization | Biopersistence concerns, potential long-term toxicity, limited biodegradability [40] |
| Hybrid Systems | High | Moderate to High | High | Synergistic functionality, enhanced targeting, improved safety profiles | Complex characterization, regulatory challenges, multi-step manufacturing [92] |
Table 2: Cost Structure Analysis for Non-Viral vs. Viral Vector Manufacturing
| Cost Factor | Non-Viral Vectors | Viral Vectors |
|---|---|---|
| Raw Materials | Synthetic chemicals, lower cost | Cell lines, plasmids, enzymes, higher cost |
| Production Time | Days to weeks | Weeks to months |
| Facility Requirements | Standard GMP facilities | Enhanced biosafety containment |
| Purification Complexity | Moderate | High (ultrafiltration, chromatography) |
| Quality Control | Standard pharmaceutical methods | Extensive adventitious agent testing |
| Overall Cost Reduction | Up to 60% compared to viral vectors | Reference standard [93] |
Key Manufacturing Complexity Factors
The production of non-viral nanoparticle vectors involves navigating multiple complexity dimensions:
- Raw Material Sourcing: Lipid nanoparticles require synthetic lipids (ionizable, structural, PEGylated, and helper lipids) with strict quality specifications [40] [9]. Polymer-based systems need medical-grade polymers with controlled molecular weights and low polydispersity [92].
- Process Parameters: Microfluidic mixing for LNP formation requires precise control of flow rates, temperature, and pH to ensure reproducible particle size and encapsulation efficiency [9].
- Quality Attributes: Critical quality attributes include particle size (50-200 nm optimal), polydispersity index (<0.2 ideal), zeta potential, encapsulation efficiency (>80% target), and sterility [40].
- Scalability Challenges: Transitioning from laboratory-scale (milliliters) to commercial production (liters to hundreds of liters) introduces mixing heterogeneity, heat transfer limitations, and purification bottlenecks [94].
Experimental Protocols
Protocol: Microfluidic Manufacturing of Lipid Nanoparticles
Objective: Reproducible preparation of siRNA/mRNA-loaded LNPs with controlled size and high encapsulation efficiency.
Materials:
- Ionizable lipid (e.g., DLin-MC3-DMA, SM-102)
- Helper lipid (DSPC)
- Cholesterol
- PEGylated lipid (DMG-PEG2000)
- Aqueous phase: 10 mM citrate buffer, pH 4.0
- Organic phase: Ethanol
- Nucleic acid payload: siRNA or mRNA in citrate buffer
- Microfluidic device (NanoAssemblr, Precision NanoSystems)
- Dialysis membranes (MWCO 100 kDa)
- Dynamic Light Scattering (DLS) instrument
Procedure:
- Lipid stock preparation: Dissolve ionizable lipid, DSPC, cholesterol, and PEG-lipid in ethanol at a molar ratio of 50:10:38.5:1.5 to a total lipid concentration of 10 mg/mL.
- Aqueous phase preparation: Dilute nucleic acid payload in 10 mM citrate buffer (pH 4.0) to a concentration of 0.1 mg/mL.
- Microfluidic mixing:
- Set total flow rate (TFR) to 12 mL/min and flow rate ratio (FRR) to 3:1 (aqueous:organic).
- Maintain temperature at 25-30°C throughout the process.
- Collect formed LNPs in a sterile container.
- Buffer exchange and purification:
- Dialyze against PBS (pH 7.4) for 4 hours at 4°C with three buffer changes.
- Optionally concentrate using centrifugal filters (100 kDa MWCO).
- Quality control:
- Measure particle size, PDI, and zeta potential by DLS.
- Determine encapsulation efficiency using Ribogreen assay.
- Confirm sterility through membrane filtration and microbiological testing.
Critical Parameters:
- Lipid and nucleic acid concentrations must be precisely controlled
- Mixing parameters (TFR, FRR) significantly impact particle characteristics
- pH affects ionizable lipid protonation and encapsulation efficiency
- Temperature control prevents lipid degradation and ensures reproducibility
Protocol: Polymeric Nanoparticle Formation via Double Emulsion
Objective: Preparation of DNA-loaded polymeric nanoparticles using PLGA or PBAEs.
Materials:
- Polymer: PLGA (50:50, acid end group, MW 10-30 kDa) or PBAEs
- Organic solvent: Dichloromethane (DML) or ethyl acetate
- Surfactant solutions: Polyvinyl alcohol (PVA) in water (1-5% w/v)
- DNA payload in TE buffer
- Probe sonicator
- Magnetic stirrer with temperature control
- Centrifugal filters
Procedure:
- Primary water-in-oil emulsion:
- Dissolve polymer in organic solvent (10 mg/mL).
- Add DNA solution (50-100 μL) to polymer solution (1 mL).
- Sonicate using probe sonicator (30% amplitude, 30 seconds on/off pulses) in ice bath.
- Secondary water-in-oil-in-water emulsion:
- Add primary emulsion to PVA solution (10 mL, 2% w/v).
- Sonicate again (20% amplitude, 45 seconds) to form double emulsion.
- Solvent evaporation:
- Stir emulsion for 3 hours at room temperature to evaporate organic solvent.
- Alternatively, use rotary evaporation under reduced pressure.
- Nanoparticle collection:
- Centrifuge at 15,000 × g for 30 minutes at 4°C.
- Wash twice with distilled water to remove excess surfactant.
- Resuspend in appropriate buffer for characterization and use.
Critical Parameters:
- Sonication time and amplitude control particle size distribution
- Polymer molecular weight and composition affect DNA release kinetics
- Surfactant concentration and type influence surface properties and stability
- Solvent removal rate impacts nanoparticle morphology
Protocol: Assessment of Scalability and Cost-Effectiveness
Objective: Systematic evaluation of manufacturing scalability and economic viability.
Materials:
- Small-scale production system (microfluidics, sonication)
- Pilot-scale equipment (tangential flow filtration, larger reactors)
- Analytical instruments (HPLC, DLS, spectrophotometry)
- Cost accounting software or spreadsheets
Procedure:
- Process scalability assessment:
- Produce nanoparticles at 10 mL, 100 mL, and 1 L scales.
- Compare critical quality attributes (size, PDI, encapsulation efficiency) across scales.
- Identify critical process parameters (CPPs) affecting product quality.
- Cost analysis:
- Calculate raw material costs per dose.
- Estimate labor and facility costs based on process time.
- Factor in purification and quality control expenses.
- Compare total cost with viral vector alternatives.
- Techno-economic modeling:
- Project costs at commercial scale (1,000+ doses per batch).
- Identify cost drivers and potential optimization strategies.
- Calculate cost of goods sold (COGS) per therapeutic dose.
Key Metrics:
- Manufacturing success rate (>80% target)
- Batch-to-batch consistency (RSD < 15%)
- Overall cost reduction compared to viral vectors (40-60% target)
- Scalability factor (maintenance of CQAs across scales)
Visualization of Manufacturing Workflows and Critical Pathways
Non-Viral Nanoparticle Manufacturing Workflow
Vector Manufacturing Complexity & Scalability
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Non-Viral Vector Development
| Reagent Category | Specific Examples | Function | Key Considerations |
|---|---|---|---|
| Ionizable Lipids | DLin-MC3-DMA, SM-102, ALC-0315 | Enable nucleic acid encapsulation and endosomal escape | pKa ~6.5 optimal for endosomal release; biodegradability reduces toxicity [9] |
| Structural Lipids | DSPC, DOPE | Stabilize lipid bilayer structure | Influence membrane fluidity and fusion capabilities [40] |
| PEGylated Lipids | DMG-PEG2000, ALC-0159 | Provide steric stabilization, reduce clearance | Shield charge, prolong circulation; but can hinder cellular uptake [40] [9] |
| Cationic Polymers | PEI, PBAEs, Chitosan derivatives | Condense nucleic acids via electrostatic interactions | Molecular weight and branching affect transfection and cytotoxicity [92] |
| Biodegradable Polymers | PLGA, PCL, PBAEs | Controlled release, reduced toxicity | Degradation rate matches therapeutic requirements [40] [92] |
| Surface Ligands | Peptides (RGD), antibodies, carbohydrates | Enable targeted delivery to specific tissues | Ligand density critical for binding avidity and internalization [92] |
| Characterization Tools | Ribogreen assay, DLS, TEM | Quantify encapsulation, size, morphology | Multiple orthogonal methods ensure accurate characterization [40] [9] |
The manufacturing landscape for non-viral nanoparticle vectors is rapidly evolving, with significant advantages emerging in scalability and cost-effectiveness compared to viral vector systems. Successful implementation requires:
Platform Process Development: Establishing standardized manufacturing platforms that can be adapted across multiple therapeutic candidates, reducing development timelines and costs [94].
Quality by Design (QbD) Approaches: Systematic understanding of how formulation and process parameters affect product quality, enabling robust manufacturing control strategies [40].
Advanced Analytics: Implementation of process analytical technologies (PAT) for real-time monitoring and control of critical quality attributes during manufacturing [94].
Supply Chain Optimization: Securing reliable sources of high-quality raw materials, particularly specialty lipids and polymers, to ensure consistent production [93].
The continued advancement of non-viral nanoparticle manufacturing technologies promises to accelerate the development of accessible gene therapies, addressing the critical challenges of production complexity, scalability, and cost that have historically limited widespread clinical application.
Analysis of Transduction Efficiency versus Transfection Efficiency
The selection between viral transduction and non-viral transfection represents a critical decision point in gene therapy and genetic research workflows. This application note provides a systematic comparison of these fundamental gene delivery approaches, with particular emphasis on their application in non-viral nanoparticle vector research. We present quantitative efficiency data, detailed protocols for both delivery strategies, and analytical frameworks for evaluating success metrics. For therapeutic applications requiring stable genomic integration, viral vectors—particularly lentiviral systems—demonstrate superior performance, while emerging non-viral nanoparticle platforms offer advantages in safety, scalability, and transient expression applications. The integration of standardized assessment protocols and appropriate technology selection directly impacts the success of gene delivery experiments and therapeutic development pipelines.
Gene delivery technologies form the foundation of modern molecular medicine, enabling revolutionary treatments for genetic disorders, cancers, and infectious diseases. The fundamental dichotomy in this field lies between transduction—utilizing viral vectors for gene transfer—and transfection—employing non-viral chemical or physical methods. While viral vectors leverage evolved biological mechanisms for highly efficient gene delivery, non-viral nanoparticle-based systems offer enhanced safety profiles and manufacturing advantages [58] [57].
The emerging paradigm in gene therapy emphasizes the importance of matching delivery technologies to specific application requirements rather than seeking universal solutions. This application note provides researchers with a structured framework for selecting and optimizing gene delivery strategies based on empirical efficiency data, practical implementation protocols, and critical quality attribute assessment. With the recent FDA approval of CRISPR/Cas9-based therapies like CASGEVY, which utilizes ex vivo electroporation, and the growing clinical adoption of lipid nanoparticles for mRNA delivery, understanding the efficiency trade-offs between different delivery platforms has never been more critical [95] [96].
Comparative Analysis of Delivery Efficiencies
Quantitative Efficiency Metrics
Gene delivery efficiency varies substantially between viral and non-viral approaches and is highly dependent on experimental parameters. The following table summarizes representative efficiency data from current literature:
Table 1: Comparative Efficiency Metrics for Gene Delivery Methods
| Delivery Method | Typical Efficiency Range | Key Applications | Critical Parameters |
|---|---|---|---|
| Lentiviral Transduction | 30-70% (immune cells) [97] | CAR-T cell engineering, stable gene expression | MOI, cell activation state, enhancers |
| Adeno-Associated Virus (AAV) Transduction | Varies by serotype and tissue | In vivo gene therapy, retinal disorders | Serotype selection, pre-existing immunity |
| Electroporation | High efficiency reported (varies by cell type) [95] | CRISPR RNP delivery, hard-to-transfect cells | Pulse parameters, cell viability optimization |
| Cationic Lipid Nanoparticles | Highly variable (10-90% depending on formulation) [96] [57] | mRNA vaccines, primary cell transfection | Lipid composition, N:P ratio, particle size |
| Polyethylenimine (PEI) | Moderate to high (cell-type dependent) [98] | Recombinant protein production, plasmid DNA delivery | Polymer molecular weight, charge ratio |
Key Characteristic Comparison
The selection between transduction and transfection involves balancing multiple vector characteristics against application requirements:
Table 2: Characteristic Comparison of Viral versus Non-Viral Delivery Systems
| Characteristic | Viral Transduction | Non-Viral Transfection |
|---|---|---|
| Mechanism | Receptor-mediated entry, natural infection pathways [58] | Endocytosis (chemical), membrane perturbation (physical) [57] [99] |
| Payload Capacity | Limited (~8kb LV, ~4.7kb AAV) [97] | Higher capacity, more flexible [96] [57] |
| Integration Profile | Stable (LV, RV) or transient (AAV, AdV) [58] | Typically transient (episomal) |
| Immunogenicity | Moderate to high [58] | Low to moderate [57] |
| Manufacturing Complexity | High, requires biosafety containment [100] | Scalable, reproducible [96] [98] |
| Regulatory Considerations | Extensive safety profiling required [58] | Generally favorable safety profile [57] |
Experimental Protocols
Protocol: Lentiviral Transduction of T Cells for CAR-T Therapy
This protocol outlines a standardized approach for transducing human T cells with lentiviral vectors, incorporating enhancements to boost transduction efficiency while maintaining cell viability and function [100] [97].
Materials and Reagents
- Activated T cells from human PBMCs (3 days post-activation with CD3/CD28 activator)
- High-titer lentiviral vector (VSV-G pseudotyped, ≥1×10⁸ IU/mL)
- Transduction medium: RPMI-1640 with 10% FBS, 2 mM L-glutamine, 100 IU/mL IL-2
- Transduction enhancers (e.g., polybrene, protamine sulfate, or commercial enhancers)
- Retronectin (optional, for pre-coating)
- Flow cytometry antibodies: CD3, CD4, CD8, viability dye
Procedure
Cell Preparation:
- Harvest activated T cells and resuspend at 1×10⁶ cells/mL in pre-warmed transduction medium.
- Assess viability using trypan blue exclusion; proceed only if viability >90%.
Vector-Cell Mixture:
- Combine cells with lentiviral vector at predetermined MOI (typically 1-10) in a minimal volume.
- Add transduction enhancer at optimal concentration (e.g., 4-8 μg/mL polybrene).
- For static transduction: Seed mixture in non-tissue culture treated 24-well plates (500 μL/well).
- For enhanced methods: Use TransB system or spinoculation (2000 × g, 32°C, 60-90 min).
Transduction Incubation:
- Incubate at 37°C, 5% CO₂ for 6-24 hours.
- For prolonged transduction, add fresh medium with cytokines after 8-12 hours.
Post-Transduction Processing:
- After 24 hours, harvest cells and centrifuge (300 × g, 5 min) to remove vector supernatant.
- Resuspend in expansion medium with IL-2 (50-100 IU/mL) at 1×10⁶ cells/mL.
- Culture for 72 hours before efficiency assessment.
Efficiency Analysis:
- Analyze transgene expression by flow cytometry using appropriate markers.
- Determine cell viability using 7-AAD or Annexin V staining.
- Quantify vector copy number (VCN) by ddPCR if needed.
Critical Process Parameters
- Cell activation state: Optimal transduction occurs 48-72 hours post-activation
- Multiplicity of Infection (MOI): Must be determined empirically for each vector batch
- Cell density: 0.5-2×10⁶ cells/mL during transduction
- Enhancer concentration: Titrate to balance efficiency and toxicity
Protocol: Lipid Nanoparticle-Mediated mRNA Transfection
This protocol describes the use of cationic lipid nanoparticles for efficient mRNA delivery, with particular relevance to CRISPR/Cas9 component delivery and vaccine development [95] [96] [57].
Materials and Reagents
- Cationic lipid nanoparticles (commercial formulations or custom-synthesized)
- mRNA cargo (e.g., Cas9 mRNA, GFP reporter mRNA)
- Serum-free cell culture medium
- Complete growth medium with serum
- Target cells (adherent or suspension)
- Transfection optimization kit (if titrating conditions)
Procedure
Nanoparticle-mRNA Complex Formation:
- Dilute mRNA in serum-free medium to 2× final concentration.
- Dilute lipid nanoparticles in equal volume of serum-free medium.
- Combine mRNA and nanoparticle solutions rapidly with gentle pipetting.
- Incubate complex at room temperature for 15-30 minutes.
Cell Preparation:
- For adherent cells: Seed 24 hours prior to achieve 60-80% confluency.
- For suspension cells: Adjust density to 0.5-1×10⁶ cells/mL on day of transfection.
Transfection:
- Remove growth medium from adherent cells, wash with PBS.
- Add nanoparticle-mRNA complexes dropwise to cells, gently swirl to distribute.
- For suspension cells: Pellet cells, resuspend in complex-containing medium.
- Incubate at 37°C, 5% CO₂ for 4-6 hours.
Post-Transfection Processing:
- Replace complex-containing medium with fresh complete medium.
- Continue incubation for 24-72 hours based on expression kinetics.
- Analyze transfection efficiency and functional outcomes.
Optimization Notes
- Charge ratio (N:P): Critical parameter affecting complex stability and efficiency
- Serum compatibility: Some formulations require serum-free conditions during transfection
- Cell health monitoring: Assess cytotoxicity 24 hours post-transfection
Visualization of Gene Delivery Pathways and Workflows
Gene Delivery Mechanism Pathways
Transduction Optimization Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Gene Delivery Research
| Reagent Category | Specific Examples | Function and Application Notes |
|---|---|---|
| Viral Vector Systems | Lentivirus (VSV-G pseudotyped), AAV (serotypes 2, 6, 9), Adenovirus (Ad5) | Delivery of genetic payload; serotype selection critical for tropism [58] [97] |
| Non-Viral Transfection Reagents | Cationic lipids (Lipofectamine, DLin-MC3-DMA), Polymers (PEI, PAMAM dendrimers) | Nucleic acid complexation and delivery; structure affects efficiency and toxicity [96] [57] [98] |
| Transduction Enhancers | Polybrene, Protamine sulfate, Retronectin, Poloxamers | Increase viral attachment and entry; concentration requires optimization [100] [97] |
| Cell Culture Supplements | IL-2, IL-7, IL-15, FBS, Human serum albumin | Maintain cell viability and function during/after genetic modification [97] |
| Analytical Tools | Flow cytometry antibodies, ddPCR reagents, viability stains (7-AAD, Annexin V) | Assessment of efficiency, cell health, and vector copy number [99] [97] |
The decision between viral transduction and non-viral transfection methodologies requires careful consideration of application-specific requirements, efficiency thresholds, and practical constraints. Viral systems, particularly lentiviral and AAV platforms, offer robust efficiency and stable expression but present challenges in manufacturing complexity and safety profiling. Non-viral nanoparticle systems, including lipid and polymeric vectors, provide advantageous safety profiles and scalability with continually improving efficiency metrics.
The emerging landscape of gene delivery emphasizes hybrid approaches that leverage the strengths of both platforms, such as nanoparticle-formulated viral vectors or virus-inspired synthetic systems. By implementing standardized protocols and rigorous assessment criteria, researchers can systematically optimize delivery strategies to advance both basic research and clinical applications. The continued refinement of these technologies promises to expand the therapeutic potential of genetic medicines across diverse disease contexts.
Gene therapy has emerged as a transformative medical intervention, modifying or manipulating genetic material within a person's cells to treat or prevent disease [86]. This approach aims to correct or replace defective genes, introduce new or modified genes, or alter gene expression to achieve therapeutic effects, primarily targeting severe conditions with limited treatment options [86]. The global viral and non-viral vector market, valued at $551.85 million in 2023, is projected to grow significantly to $3.505 billion by 2030 at a robust CAGR of 31.84% [101], reflecting the intense innovation in this sector.
Vectors serve as the critical delivery vehicles for therapeutic genetic material, with both viral and non-viral platforms offering distinct advantages and limitations [14] [86]. The selection between these platforms involves careful consideration of vector safety, target efficiency, and commercial feasibility [85]. To date, 35 vector-based gene therapies have received market approval globally, with 29 utilizing viral vectors and 6 employing non-viral approaches [86] [1]. This article examines the current state, applications, and future trajectory of both viral and non-viral gene delivery systems within the context of advancing non-viral nanoparticle vector research.
Current Market and Clinical Landscape
The cell and gene therapy market is experiencing substantial growth, with projections indicating it will exceed $70 billion globally over the next decade [94]. This expansion is being driven by maturing pipelines that are expanding beyond rare diseases into oncology, neurology, and chronic conditions, coupled with an accelerating pace of regulatory approvals [94]. Manufacturing demand has risen sharply to support a doubling of clinical trials since 2019, with more than 10 new commercial products approved in recent years [94].
The viral and non-viral vector manufacturing market specifically has experienced rapid expansion, projected to increase from $8.67 billion in 2024 to $10.28 billion by 2025, reflecting an impressive compound annual growth rate of 18.5% [102]. Exponential expansion is projected to continue, with the market reaching $22.01 billion by 2029, fueled by a CAGR of 21.0% [102]. This growth is fundamentally driven by escalating demand for gene therapies, widening applications for rare ailments, increased capital allocation toward biomanufacturing facilities, and the growing integration of personalized medicine approaches [102].
Approved Gene Therapy Products
Table 1: Approved Viral Vector-Based Gene Therapies (Selected Examples)
| Drug Name | Vector Type | Condition | Year First Approved | Regulatory Agency |
|---|---|---|---|---|
| Kymriah | LV (ex vivo) | Acute lymphocytic leukaemia; lymphoma | 2017 | FDA |
| Zolgensma | AAV | Spinal muscular atrophy | 2019 | EMA |
| Luxturna | AAV | Leber congenital amaurosis; retinitis pigmentosa | 2017 | FDA/EMA |
| Gendicine | Ad | Head and neck cancer | 2003 | CFDA |
| Skysona | LV (ex vivo) | Early cerebral adrenoleukodystrophy | 2021 | FDA |
| Strimvelis | RV | Adenosine deaminase deficiency | 2016 | EMA |
| Adstiladrin | Ad | Bladder cancer | 2022 | FDA |
| Hemgenix | AAV | Hemophilia B | 2022 | FDA |
| Vyjuvek | HSV | Dystrophic epidermolysis bullosa | 2023 | FDA |
| Lyfgenia | LV (ex vivo) | Sickle cell disease | 2023 | FDA |
Table 2: Approved Non-Viral Vector-Based Gene Therapies
| Drug Name | Vector Type | Condition | Year First Approved | Regulatory Agency |
|---|---|---|---|---|
| Onpattro | LNP | Human transthyretin amyloidosis (hATTR) | 2018 | FDA |
| Givlaari | GalNAc | Acute hepatic porphyria (AHP) | 2019 | FDA |
| Oxlumo | GalNAc | Primary hyperoxaluria type1 (PH1) | 2020 | FDA |
| Leqvio | GalNAc | Hypercholesterolaemia | 2020 | FDA |
| Amvuttra | GalNAc | hATTR | 2022 | FDA |
| Rivfloza | GalNAc | Primary hyperoxaluria | 2023 | FDA |
Viral Vector Platforms: Current Workhorses of Gene Therapy
Viral vectors have been used as gene therapy vehicles since 1975 due to their high infection efficiency in vivo and diversity of targeted tissues [86]. They remain the primary gene delivery vectors today, with lentiviruses (LV), adenoviruses (Ad), and adeno-associated viruses (AAV) being the most widely used, accounting for over 80% of approved viral-based gene therapy products [86]. These vectors capitalize on viruses' natural ability to enter cell nuclei and deliver genetic material [85].
Key Viral Vector Characteristics and Applications
Table 3: Comparative Analysis of Major Viral Vector Platforms
| Parameter | Adeno-Associated Virus (AAV) | Lentivirus (LV) | Adenovirus (Ad) |
|---|---|---|---|
| Payload Capacity | ~4.7 kb [1] | Limited [85] | Large (up to 12 kb) [85] |
| Integration | Non-integrating (episomal) [85] | Integrating [85] | Non-integrating (episomal) |
| Cell Targeting | Dividing and non-dividing cells [85] | Dividing and non-dividing cells [85] | Primarily dividing cells |
| Gene Expression | Long-term but transient in dividing cells [85] | Long-term (passed to daughter cells) [85] | Transient |
| Immunogenicity | Low [85] [1] | Moderate | High [1] |
| Primary Applications | In vivo therapies (e.g., Luxturna, Zolgensma) [1] | Ex vivo therapies (CAR-T, stem cells) [1] | Cancer therapy, vaccines [1] |
| Key Advantages | Multiple serotypes for tissue specificity; good safety profile [85] [1] | Stable long-term expression; broad tropism [85] | High transduction efficiency; large cargo capacity [85] [1] |
| Key Limitations | Limited cargo size; pre-existing immunity [1] | Risk of insertional mutagenesis [1] | Strong immune response [1] |
Viral Vector Manufacturing Challenges and Innovations
Viral vector manufacturing remains a major bottleneck: complex, inefficient, and prohibitively expensive [103]. The inherent complexity of viral vector-based therapies represents a primary barrier to commercializing cost-effective cell and gene therapies [103]. Unlike traditional biologics, these therapies consist of intricate components—a genetic payload encased within a protein capsid (with LV and RV vectors further enveloped in a lipid membrane)—that must assemble carefully and function synergistically [103].
Manufacturing challenges are compounded by the urgency to accelerate development for patients with severe conditions, leading to suboptimal scaling of early-stage processes [103]. Standardization remains elusive with no universally adopted production platform, and processes vary widely between vector types and even across serotypes within the same class [103]. Downstream processing presents similar challenges, with purification often involving several sequential steps—affinity capture, anion-exchange polishing chromatography, ultracentrifugation, and tangential-flow filtration—tailored to specific vectors and resulting in poor recovery rates [103].
Innovations addressing these challenges include:
- Synthetic DNA: Replacing plasmid DNA produced through bacterial fermentation with enzymatically produced synthetic DNA to eliminate bacterial contaminants, shorten production timelines, and reduce costs [103].
- Stable Producer Cell Lines: Shifting from transient transfection to packaging and producer cell lines that require plasmid DNA only during initial development, then stably express necessary viral components [103].
- Fixed-Bed Bioreactors: Implementing closed, automated fixed-bed bioreactor systems for adherent cell culture (particularly for LV vectors) to reduce labor costs, facility footprints, and improve vector yield consistency [103].
Non-Viral Vector Platforms: Emerging Alternatives
Non-viral vectors have gained momentum as safer, more scalable alternatives to viral platforms [1]. Unlike viruses, they do not integrate into the host genome or trigger strong immune responses, and their broader cargo capacity and lower production cost make them attractive for diverse applications [8] [1]. While non-viral vectors must overcome challenges like lower transfection efficiency, ongoing research is rapidly addressing these limitations [8] [85].
Key Non-Viral Vector Platforms
Lipid Nanoparticles (LNPs)
Lipid nanoparticles have gained widespread recognition following their successful use in mRNA COVID-19 vaccines [1]. In gene therapy, LNPs deliver siRNA and CRISPR components [1]. Patisiran (Onpattro), approved in 2018 for hereditary transthyretin amyloidosis, was the first LNP-based siRNA therapy [86] [1]. LNPs form when positively charged cationic lipids create electrostatic interactions with negatively charged genetic material, forming lipoplexes [85]. Recent advances include modified lipids with positive charges to form lipoplexes with DNA and escape endosomal vesicles, but with neutral charges at physiological pH to enable delivery [85].
N-Acetylgalactosamine (GalNAc) Conjugates
The GalNAc platform enables liver-targeted delivery of RNA therapies through subcutaneous administration [1]. GalNAc conjugation has enabled multiple FDA-approved drugs, including Givlaari, Oxlumo, and Leqvio, to effectively treat rare genetic and cardiovascular diseases [86] [1]. This approach exploits the high expression of asialoglycoprotein receptors on hepatocytes, facilitating efficient receptor-mediated uptake of GalNAc-conjugated therapeutics [86].
Polymeric and Inorganic Vectors
Cationic polymers like polyethylenimine (PEI) represent another major non-viral vector category, offering versatile chemical structures with high capacity for genetic material [85]. PEI demonstrates the greatest transfection efficiency among non-viral vectors, with its positive charge generating an osmotic effect to induce endosome burst and assist transfection efficiency [85]. However, concerns about cytotoxicity due to non-biodegradability have driven development of biodegradable alternatives like PBAEs and PLAs [85]. Inorganic materials such as silica nanoparticles, gold nanoparticles, magnetic nanoparticles, and carbon nanotubes offer exceptional stability and are being explored for specialized applications [85].
Protocol: Optimizing PEI-Based Gene Delivery to T Cells
Recent research has demonstrated comprehensive optimization of PEI-mediated gene delivery to human T cells, which holds promise for reducing the cost and complexity of preparing engineered T cells [22]. The following protocol details this optimized methodology:
Principle: Fine-tuning characteristics of PEI/DNA nanoparticles, culture conditions, cellular physiology, and transfection protocols to enhance gene delivery into T cells [22].
Materials:
- Branched polyethylenimine (PEI, molecular weight ~25,000 Da)
- Plasmid DNA encoding gene of interest
- RPMI 1640 culture medium
- Human T cells
- Transfection vials
Procedure:
Nanoparticle Formation:
Transfection Setup:
- Perform reverse transfection in vials rather than conventional direct transfection in culture plates (20-fold increase in cellular uptake and transfection efficiency) [22].
- Increase cell seeding density to boost PEI-mediated gene delivery [22].
- Add complete media shortly after transfection to enhance outcomes [22].
Cellular Physiology Modulation:
Analysis:
- Assess transfection efficiency using appropriate methods (e.g., flow cytometry for reporter gene expression).
- Evaluate cytotoxicity using standardized assays (e.g., MTT, LDH release).
Notes: This optimized approach significantly enhances gene delivery efficiency while maintaining cell viability, potentially accelerating the development of immune cell therapies for human diseases [22].
Diagram 1: PEI-Mediated T Cell Transfection Workflow
Comparative Analysis: Strategic Selection Criteria
The choice between viral and non-viral vectors depends on various factors including the specific therapeutic goal, target cell type, duration of gene expression, safety profile, and immune response [86]. Each platform presents unique advantages and limitations that must be weighed for each application.
Advantages and Limitations Comparison
Table 4: Strategic Selection Criteria for Gene Delivery Platforms
| Consideration | Viral Vectors | Non-Viral Vectors |
|---|---|---|
| Transfection Efficiency | High transduction efficiency [85] | Lower gene transfer efficiency [85] |
| Safety Profile | Immune response concerns; insertional mutagenesis risk (LV, RV) [85] [1] | Superior safety profile; low immunogenicity and mutagenesis risk [8] [85] |
| Manufacturing Scalability | Complex, high-cost manufacturing; scalability challenges [85] [103] | Easier manufacturing; better commercial scalability [85] |
| Payload Capacity | Limited (especially AAV) [1] | Large cargo capacity [8] [85] |
| Targeting Specificity | Tissue-specific tropism (especially AAV serotypes) [85] | Lower specificity; off-target biodistribution concerns [85] |
| Regulatory Precedent | Extensive clinical history; 29 approved therapies [86] | Growing but limited approval history (6 approved therapies) [86] |
| Cost Considerations | High production costs [85] [103] | Lower manufacturing costs [85] |
The Scientist's Toolkit: Essential Research Reagents
Table 5: Key Research Reagent Solutions for Gene Delivery Research
| Reagent/Material | Function | Example Applications |
|---|---|---|
| PEI (Polyethylenimine) | Cationic polymer for DNA complexation; facilitates endosomal escape [85] [22] | Non-viral gene delivery to T cells and other hard-to-transfect cells [22] |
| Cationic Lipids | Form lipoplexes with nucleic acids; component of LNPs [85] | siRNA delivery (Onpattro); mRNA vaccine delivery [1] |
| GalNAc Conjugates | Liver-targeted delivery via asialoglycoprotein receptor binding [1] | RNAi therapeutics for hepatic diseases (Givlaari, Oxlumo) [86] [1] |
| AAV Serotypes | Engineered viral capsids with tissue-specific tropism [85] [1] | In vivo gene therapy for retinal, muscular, and neurological disorders [14] [1] |
| Lentiviral Packaging Systems | Production of replication-incompetent lentiviral vectors [86] | Ex vivo cell engineering (CAR-T cells, hematopoietic stem cells) [86] [1] |
| CRISPR-Cas9 Components | Gene editing machinery requiring efficient delivery systems [8] | Therapeutic gene editing when combined with viral or non-viral delivery [8] [1] |
Future Perspectives and Research Directions
The future of gene delivery platforms is evolving toward technological diversification, with multiple platforms coexisting as developers strategically select approaches based on indication, therapeutic delivery route, and manufacturing feasibility [94]. Several key trends are shaping this future trajectory.
Manufacturing Innovation and Scalability
The industry is shifting toward automated, digital, and decentralized manufacturing models to improve production speed, efficiency, quality, and global access [94]. Automated and closed manufacturing systems, particularly for autologous therapies, are transforming cell and gene therapy from artisanal processes to industrialized platforms [94]. Digital tools and AI are alleviating production bottlenecks, streamlining production, and enhancing quality assurance processes [94]. These innovations directly address quality control testing, historically one of the largest bottlenecks in manufacturing [94].
The role of contract development and manufacturing organizations (CDMOs) is evolving from service provider to innovation partner as leading organizations invest in new capacity, develop new competencies, and proactively anticipate future requirements [94]. This partnership model enables smaller innovators to access advanced manufacturing capabilities and regulatory expertise previously available only to larger organizations [94].
Technology Convergence and Platform Evolution
Several technological synergies are driving the next generation of gene delivery platforms:
Hybrid Vector Systems: Combining advantageous elements of both viral and non-viral systems to create novel delivery platforms with improved safety and efficiency profiles.
CRISPR Integration: The integration of CRISPR gene-editing tools with both viral and non-viral vector systems is enhancing precision and opening new avenues in personalized medicine [101]. NTLA-2002 has already demonstrated the feasibility of LNP-mediated CRISPR delivery for hereditary angioedema [1].
Novel Administration Strategies: Research is advancing alternative delivery methods beyond systemic injection. Localized approaches (subretinal, intravitreal, round window membrane, PSCC routes for inner ears, intracerebroventricular, and intraparenchymal for brain) reduce required doses and decrease severe immune response risks, though challenges remain with invasive administration and uneven distribution [86].
In Vivo Cell Engineering: Growing interest in in vivo CAR-T and in vivo gene editing approaches that bypass complex ex vivo cell manipulation, potentially offering easier administration, lower cost, and greater scalability [94].
Diagram 2: Future Gene Delivery Platform Trajectory
The evolving roles of viral and non-viral platforms reflect a dynamic gene therapy landscape moving toward personalized, scalable, and accessible treatments. Viral vectors currently dominate approved therapies, with AAV and LV platforms enabling remarkable clinical successes across diverse disease areas [14] [86] [1]. However, manufacturing complexities, immunogenicity concerns, and payload limitations continue to drive innovation in viral vector engineering and production technologies [103].
Non-viral vectors, particularly lipid nanoparticles and GalNAc conjugates, are gaining substantial momentum with demonstrated clinical success and compelling advantages in safety, manufacturing scalability, and payload flexibility [8] [1]. While challenges remain in transfection efficiency and tissue-specific targeting, ongoing research is rapidly addressing these limitations through novel materials, formulations, and delivery strategies [85] [22].
The future trajectory points toward platform diversification rather than consolidation, with viral and non-viral systems coexisting and complementing each other in an expanding therapeutic toolkit [94]. The convergence of these delivery technologies with gene editing tools like CRISPR, coupled with manufacturing innovations in automation and digitalization, will ultimately enable broader application of gene therapies beyond rare diseases to oncology, cardiovascular conditions, neurological disorders, and chronic diseases [94] [1] [101]. As the field advances, strategic selection and continued optimization of both viral and non-viral platforms will be essential to fully realize the transformative potential of gene therapy.
Conclusion
Non-viral nanoparticle vectors represent a transformative advancement in gene therapy, successfully addressing critical limitations of viral vectors, including immunogenicity, cargo constraints, and manufacturing complexity. The progression of lipid-based, polymer-based, and inorganic systems has enabled clinical validation, evidenced by approved products and a robust pipeline targeting genetic, oncological, and neurological disorders. However, the path forward requires intensified research to overcome persistent hurdles in transfection efficiency, precise extrahepatic targeting, and long-term expression stability. The future of gene delivery does not necessarily hinge on the supremacy of one platform over the other, but rather on the strategic application of both viral and non-viral vectors based on specific therapeutic contexts. Continued innovation in material science, surface engineering, and formulation will undoubtedly expand the therapeutic reach of non-viral nanoparticles, solidifying their role in making gene therapy a mainstream treatment modality for a broader range of human diseases.
References
- 1. The State and Future of Viral, Nonviral Vectors for Gene ... [https://www.ajmc.com/view/the-state-and-future-of-viral-non-viral-vectors-for-gene-therapy]
- 2. Progress and challenges in viral vector manufacturing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4802372/]
- 3. Modeling integration site data for safety assessment with ... [https://www.nature.com/articles/s41467-025-63017-w]
- 4. Characterization of AAV vectors: A review of analytical ... [https://www.sciencedirect.com/science/article/pii/S2329050124001256]
- 5. Synergistic enhancement of AAV gene delivery in 2D cells ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12617951/]
- 6. Synergistic enhancement of AAV gene delivery in 2D cells ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0336164]
- 7. Systematic review and meta-analysis of the effectiveness of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12603288/]
- 8. Non-Viral Gene Delivery Systems: Current Advances and ... [https://pubmed.ncbi.nlm.nih.gov/40874945/]
- 9. Advances in Nanoparticles as Non-Viral Vectors for ... [https://www.mdpi.com/1999-4923/16/9/1197]
- 10. Advances in Gene Delivery Systems - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC3245684/]
- 11. CRISPR Delivery Methods: Cargo, Vehicles, and Challenges [https://www.synthego.com/blog/delivery-crispr-cas9/]
- 12. Gene Delivery Systems | NIST [https://www.nist.gov/programs-projects/gene-delivery-systems]
- 13. Gene Delivery Systems Market Growth Statistics - 2035 [https://www.factmr.com/report/gene-delivery-systems-market]
- 14. Viral and non-viral vectors in gene therapy: current state ... [https://www.sciencedirect.com/science/article/pii/S2352396425002786]
- 15. Optimizing the targeting of lipid nanoparticles for gene therapy [https://pubs.rsc.org/en/content/articlehtml/2025/nh/d5nh00351b]
- 16. Overview of the non-viral gene delivery methods [https://insidetx.com/resources/reviews/overview-of-the-non-viral-gene-delivery-methods/]
- 17. Nanotechnology-based non-viral vectors for gene delivery ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2024.1349077/full]
- 18. Recent advances in surface decoration of nanoparticles ... [https://www.frontiersin.org/journals/nanotechnology/articles/10.3389/fnano.2024.1456939/full]
- 19. Cationic lipids via multi-component Passerini reaction for ... [https://pubs.rsc.org/doi/d3nj05949a]
- 20. Quantification of Cellular and Nuclear Uptake Rates ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5061650/]
- 21. Review Uptake mechanisms of non-viral gene delivery [https://www.sciencedirect.com/science/article/abs/pii/S0168365911009473]
- 22. Enhancing non-viral gene delivery to human T cells ... [https://pubmed.ncbi.nlm.nih.gov/39837215/]
- 23. New Color-Coded Test Quickly Reveals If Medical ... [https://jhtie.jhmi.edu/news/new-color-coded-test-quickly-reveals-if-medical-nanoparticles-deliver-their-payload/]
- 24. Quantitative Comparison of Tumor Delivery for Multiple ... [https://www.nature.com/articles/srep05840]
- 25. Harnessing 3D cell models and high-resolution imaging to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12277333/]
- 26. Visualizing lipid nanoparticle trafficking for mRNA vaccine ... [https://www.sciencedirect.com/science/article/pii/S1525001625000127]
- 27. Advancing gene editing: the role of lipid nanoparticles in ... [https://www.drugtargetreview.com/article/188056/advancing-gene-editing-the-role-of-lipid-nanoparticles-in-crispr-delivery/]
- 28. Design Strategies for Novel Lipid Nanoparticle for mRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12497691/]
- 29. Lipid Nanoparticles: A Breakthrough in CRISPR Delivery ... [https://www.genscript.com/lipid-nanoparticles-the-vanguard-of-crispr-delivery-systems.html]
- 30. Advances in RNA-based therapeutics: current breakthroughs ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12592931/]
- 31. Preclinical testing of antiviral siRNA therapeutics delivered in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12507954/]
- 32. Landscape of small nucleic acid therapeutics [https://www.nature.com/articles/s41392-024-02112-8]
- 33. Lung and liver editing by lipid nanoparticle delivery of a ... [https://www.nature.com/articles/s41587-024-02437-3]
- 34. Enhancing RNA-lipid nanoparticle delivery: Organ [https://www.sciencedirect.com/science/article/pii/S0168365924005765]
- 35. Advances in RNA therapeutics: Classes, innovations and ... [https://www.sciencedirect.com/science/article/pii/S3050632825000162]
- 36. A simple epoxide modification strategy to construct amino ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc04801j]
- 37. Synthesis, characterization, and evaluation of low molecular ... [https://pubs.rsc.org/en/content/articlehtml/2025/na/d5na00169b]
- 38. Polymer-based coating of adeno-associated viral particles ... [https://www.sciencedirect.com/science/article/pii/S0168365925005164]
- 39. The Next Generation of Transfection Polymers [https://blog.curapath.com/the-next-generation-of-transfection-polymers]
- 40. Current advances in non-viral nanoparticle-based gene ... [https://www.sciencedirect.com/science/article/abs/pii/S1773224724007524]
- 41. Medical importance and pharmacokinetics of gold ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12512626/]
- 42. Research Progress on Nucleus-Targeting Carbon ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12335257/]
- 43. Top Gold Nanoparticle Trends in 2025: Tiny Particles, Big ... [https://www.torskal.com/the-future-is-gold-and-tiny-top-trends-in-gold-nanoparticles-research-in-2025/?srsltid=AfmBOorjJI3cXmSiDmGPRzZ019mtzO1NXZRonGXkf2JE0O3q3P4n1NzP]
- 44. What's The State Of Gold Nanoparticles (AuNPs) Materials ... [https://briandcolwell.com/whats-the-state-of-gold-nanoparticles-aunps-materials-innovation-in-2025/]
- 45. Green synthesized mesoporous silica nanoparticles offer a ... [https://pubs.rsc.org/en/content/articlehtml/2025/ra/d5ra06132f?page=search]
- 46. Carbon Nanotubes as Excellent Adjuvants for Anticancer ... [https://www.mdpi.com/2073-4409/14/14/1052]
- 47. Nanoparticle-mediated gene delivery techniques in plant ... [https://www.frontiersin.org/journals/nanotechnology/articles/10.3389/fnano.2025.1516180/full]
- 48. Extracellular Vesicles: A New Star for Gene Drug Delivery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10924919/]
- 49. Exosome-Based Drug Delivery: A Next-Generation Platform ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12567338/]
- 50. Extracellular vesicles for targeted drug delivery [https://pmc.ncbi.nlm.nih.gov/articles/PMC12486671/]
- 51. Viral vectors and extracellular vesicles: innate delivery ... [https://www.nature.com/articles/s41417-023-00597-z]
- 52. Extracellular Vesicles and Nanoparticles in Regenerative and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12566876/]
- 53. Extracellular vesicle-based drug overview: research ... [https://www.nature.com/articles/s41392-025-02312-w]
- 54. : a review of biologic function, diagnostic and targeted... Exosomes [https://jbiomedsci.biomedcentral.com/articles/10.1186/s12929-024-01055-0]
- 55. Recent Progress and Challenges in Exosomes for ... [https://www.sciencedirect.com/science/article/abs/pii/S3050475925007778]
- 56. Application of Non-Viral Vectors in Drug Delivery and Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8512075/]
- 57. Emerging non-viral vectors for gene delivery [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-023-02044-5]
- 58. Viral and Non-Viral Systems to Deliver Gene Therapeutics ... [https://www.mdpi.com/1422-0067/25/13/7333]
- 59. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]
- 60. 100 cell and gene therapy leaders to watch in 2025 [https://www.drugdiscoverytrends.com/100-cell-and-gene-therapy-leaders-to-watch-in-2025/]
- 61. ChristianaCare Gene Editing Institute Achieves CRISPR ... [https://www.biospace.com/press-releases/christianacare-gene-editing-institute-achieves-crispr-breakthrough-that-reverses-chemotherapy-resistance-in-lung-cancer]
- 62. Emerging non-viral vectors for gene delivery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10433663/]
- 63. Endosomal escape mechanisms of extracellular vesicle ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11648457/]
- 64. Targeted degradation of cell surface proteins through ... [https://www.nature.com/articles/s41467-025-62776-w]
- 65. Enhancing non-viral DNA delivery systems [https://www.sciencedirect.com/science/article/pii/S0168365924008459]
- 66. Delivering the Message: Translating mRNA Therapy for Liver ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12361847/]
- 67. Reformulating lipid nanoparticles for organ-targeted mRNA ... [https://www.nature.com/articles/s41467-024-50093-7]
- 68. Formulation-Driven Optimization of PEG-Lipid Content in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12388858/]
- 69. Overcoming cellular barriers for viral and nonviral gene ... [https://pubmed.ncbi.nlm.nih.gov/40112901/]
- 70. Quantitative comparison of intracellular trafficking and ... [https://pubmed.ncbi.nlm.nih.gov/16364692/]
- 71. Advancing cancer gene therapy: the emerging role of ... [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-025-03433-8]
- 72. Nanodelivery of nucleic acids [https://www.nature.com/articles/s43586-022-00104-y]
- 73. A Review on the Stability Challenges of Advanced Biologic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12114724/]
- 74. Pharmacometric modeling of lipid nanoparticle-encapsulated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12418826/]
- 75. The Impact of QSP Modeling on the Design ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12625082/]
- 76. Bringing New Ideas to AAV Gene Therapy [https://themedicinemaker.com/issues/2025/articles/november/bringing-new-ideas-to-aav-gene-therapy/]
- 77. Theoretical Models and Simulations of Gene Delivery with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11939958/]
- 78. Smart and Innovative Nanoparticle Systems [https://www.saspublishers.com/article/22680/]
- 79. Advanced rational design of polymeric nanoparticles for ... [https://www.sciencedirect.com/science/article/abs/pii/S0168365925005607]
- 80. Data-driven optimization of nanoparticle size using the ... [https://pubs.rsc.org/en/content/articlehtml/2025/nr/d5nr01664a]
- 81. Drug Delivery Technologies Impacting 2025 and Beyond [https://www.pharmaadvancement.com/pharma-trends/drug-delivery-technologies-impacting-2025-and-beyond/]
- 82. Bridging the Gap: The Role of Advanced Formulation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12579833/]
- 83. Comparison of Drug Delivery Systems with Different Types of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11679882/]
- 84. Quantifying nanoparticle delivery: challenges, tools, and ... [https://www.sciencedirect.com/science/article/abs/pii/S0958166923001520]
- 85. Viral vs Non-Viral Vectors in Gene Therapy [https://www.phacilitate.com/insights-resources/gene-therapy-big-debate-viral-vectors-vs-non-viral-vectors]
- 86. Viral and non-viral vectors in gene therapy: current state and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12271757/]
- 87. Immunogenicity of lipid nanoparticles and its impact on the ... [https://www.nature.com/articles/s12276-023-01086-x]
- 88. The next frontier of in vivo gene therapy: are LNPs the new ... [https://www.darkhorseconsultinggroup.com/post/the-next-frontier-of-in-vivo-gene-therapies]
- 89. Beyond the Virus: How Non-Viral Vectors Are Changing Gene ... [https://us.progen.com/post/Beyond-the-Virus-How-Non-Viral-Vectors-Are-Changing-Gene-Therapy]
- 90. Preparation and Characterization of Lipid Nanoparticles ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12457836/]
- 91. Modulation of lipid nanoparticle-formulated plasmid DNA ... [https://www.sciencedirect.com/science/article/pii/S2666379125001089]
- 92. Macromolecular gene delivery systems: Advancing non- ... [https://www.eurekalert.org/news-releases/1095020]
- 93. Gene Therapy Market Size, Growth Analysis (2025-2030) [https://www.mordorintelligence.com/industry-reports/gene-therapy-market]
- 94. Trends Shaping the Future of Cell and Gene Therapy ... [https://www.agcbio.com/biopharma-blog/trends-shaping-the-future-of-cell-and-gene-therapy-manufacturing]
- 95. CRISPR/Cas9 Delivery Systems to Enhance Gene Editing ... [https://www.mdpi.com/1422-0067/26/9/4420]
- 96. Transfection Technologies for Next-Generation Therapies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12347970/]
- 97. Optimizing viral transduction in immune cell therapy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12046913/]
- 98. Macromolecular Gene Delivery Systems: Advancing Non- ... [https://www.xiahepublishing.com/2572-5505/JERP-2025-00009]
- 99. Mapping cellular processes that determine delivery of plasmid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11969153/]
- 100. a novel transduction device for efficient and scalable gene ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06836-1]
- 101. Viral and Non-Viral Vector Market Analysis (2025-2031) [https://www.linkedin.com/pulse/viral-non-viral-vector-market-analysis-2025-2031-fogee]
- 102. 2025-2034 Viral And Non-Viral Vector Manufacturing Market [https://www.openpr.com/news/4272467/2025-2034-viral-and-non-viral-vector-manufacturing-market]
- 103. Transformative Advances in Viral Vector Manufacturing [https://www.pharmasalmanac.com/articles/transformative-advances-in-viral-vector-manufacturing-unlocking-commercial-scalability-consistency-and-cost-effectiveness]