Polymeric Nanoparticles vs. Liposomal Formulations: A Comprehensive Analysis for Advanced Drug Delivery
This article provides a systematic comparison of polymeric nanoparticles (PNPs) and liposomal formulations, two cornerstone technologies in modern nanomedicine.
Polymeric Nanoparticles vs. Liposomal Formulations: A Comprehensive Analysis for Advanced Drug Delivery
Abstract
This article provides a systematic comparison of polymeric nanoparticles (PNPs) and liposomal formulations, two cornerstone technologies in modern nanomedicine. Tailored for researchers, scientists, and drug development professionals, it explores the fundamental structures, synthesis methods, and material sciences underpinning each system. The analysis delves into their respective applications in overcoming biological barriers, such as the blood-brain barrier and tumor microenvironment, and details advanced engineering strategies for enhancing targeting, circulation time, and controlled release. A critical evaluation of scalability, stability, and regulatory challenges is presented, alongside a comparative assessment of their clinical translation, therapeutic efficacy, and performance in specific disease contexts. This review synthesizes key decision-making criteria for selecting the optimal nanocarrier and discusses future trajectories, including hybrid systems and personalized medicine approaches.
Deconstructing the Architectures: Core Structures and Material Sciences of PNPs and Liposomes
Polymeric nanoparticles (PNPs) represent a cornerstone of modern nanomedicine, offering innovative solutions to complex drug delivery challenges. These nanoscale carriers, typically ranging from 10 to 1000 nanometers, are engineered to transport therapeutic agents to specific sites in the body, thereby enhancing drug efficacy while minimizing systemic side effects [1]. Their nanoscale dimensions facilitate cellular uptake and allow these particles to cross biological barriers, including the challenging blood-brain barrier [1]. A major advantage of PNPs is their chemical versatility, enabling the creation of a virtually limitless range of polymers with tailored properties through various polymerization methods and functional groups [1].
Biodegradability is a critical characteristic of modern polymeric nanoparticles, distinguishing them from earlier non-degradable materials. Biodegradable polymeric nanoparticles (BPNPs) are designed to break down into non-toxic byproducts that are safely eliminated from the body, minimizing long-term accumulation and toxicity concerns [2]. This review focuses on two of the most prominent biodegradable polymers in pharmaceutical applications: poly(lactic-co-glycolic acid) (PLGA), a synthetic polymer, and chitosan, a natural polysaccharide. Through a comprehensive comparison of their properties, applications, and performance data, this guide provides researchers with evidence-based insights for selecting appropriate polymer systems for specific drug delivery challenges within the broader context of nanoparticle development, particularly in comparison to liposomal formulations.
Composition and Structure of Polymeric Nanoparticles
Polymeric nanoparticles are classified based on their structural organization into two primary categories: nanospheres and nanocapsules. Nanospheres are matrix-like systems where the drug is uniformly dispersed throughout the polymer matrix, while nanocapsules exhibit a reservoir system with a drug-filled core surrounded by a polymeric shell [2]. This structural distinction significantly influences drug loading capacity, release kinetics, and protection of the encapsulated therapeutic agent.
The composition of PNPs can be derived from either natural or synthetic sources. Natural polymers include polysaccharides such as chitosan, alginate, and cellulose, as well as polypeptides like gelatin, collagen, and albumin [2]. These materials are characterized by excellent biocompatibility and often inherent bioactivity. Synthetic polymers, particularly PLGA and other polyesters like polylactic acid (PLA) and poly-ε-caprolactone (PCL), offer precisely controlled properties including degradation rates, mechanical strength, and drug release profiles [2]. The selection between natural and synthetic polymers depends on the specific application requirements, with increasing research focused on hybrid systems that combine the advantages of both categories.
Table 1: Classification of Biodegradable Polymers for Nanoparticle Formulation
| Polymer Type | Examples | Sources | Key Characteristics |
|---|---|---|---|
| Natural Polymers | Chitosan [2] | Crustacean shells [3] | Mucoadhesive, biocompatible, antimicrobial |
| Alginate [2] | Brown algae | pH-responsive gelling, biocompatible | |
| Gelatin [2] | Animal collagen | Low immunogenicity, tunable isoelectric point | |
| Albumin [2] | Human blood, plants | Low immune reactivity, high binding capacity | |
| Synthetic Polymers | PLGA [4] [2] | Lactide and glycolide monomers | Tunable degradation, controlled release, FDA-approved |
| PLA [2] | Lactic acid monomers | Slower degradation than PLGA, high mechanical strength | |
| PCL [2] | ε-Caprolactone monomers | Slow degradation, suitable for long-term delivery |
Key Characteristics of Polymeric Nanoparticles
Biodegradability and Biocompatibility
Biodegradability is a fundamental requirement for polymeric nanoparticles intended for therapeutic use. PLGA undergoes hydrolysis in the body, breaking down into its monomeric constituents—lactic acid and glycolic acid—which enter the Krebs cycle and are metabolized into water and carbon dioxide [2]. This degradation rate can be precisely tuned by varying the lactide to glycolide ratio and molecular weight, with 50:50 ratios typically degrading fastest [4]. Chitosan, in contrast, is primarily degraded by lysozymes through enzymatic hydrolysis of its glycosidic linkages, producing non-toxic oligosaccharides that are incorporated into metabolic pathways [2]. Both polymers have demonstrated excellent safety profiles, with PLGA having multiple FDA-approved products and chitosan classified as Generally Recognized as Safe (GRAS) by the FDA [3].
Drug Loading and Release Kinetics
The drug encapsulation efficiency and release profiles of PNPs are influenced by multiple factors including polymer composition, drug-polymer interactions, and nanoparticle structure. PLGA nanoparticles exhibit high drug encapsulation efficiency, particularly for hydrophobic compounds, protecting encapsulated drugs from degradation and enhancing stability [2]. The controlled release capability of PLGA is one of its most valued attributes, with release profiles that can extend from days to several months depending on formulation parameters [4]. Chitosan nanoparticles demonstrate particularly high efficiency for encapsulating macromolecules like proteins and peptides, with their release kinetics influenced by the polymer's mucoadhesive properties and swelling behavior in different physiological environments [2].
Comparative Analysis of PLGA and Chitosan Nanoparticles
Table 2: Comprehensive Comparison of PLGA and Chitosan Nanoparticles
| Parameter | PLGA Nanoparticles | Chitosan Nanoparticles |
|---|---|---|
| Polymer Origin | Synthetic copolymer [4] | Natural polysaccharide from chitin [3] |
| Biodegradation | Hydrolysis into lactic/glycolic acids [2] | Enzymatic degradation by lysozymes [2] |
| Biocompatibility | Excellent; FDA-approved for multiple products [4] | Excellent; GRAS status by FDA [3] |
| Drug Release Profile | Sustained release (weeks to months) [4] | Variable release (hours to days) [2] |
| Mucoadhesiveness | Low without modification [2] | High; adheres to mucosal surfaces [2] |
| Antimicrobial Activity | Not inherent | Inherent antimicrobial properties [2] |
| Typical Size Range | 50-300 nm [5] | 80-400 nm [2] |
| Loading Efficiency | High for hydrophobic drugs [2] | High for macromolecules [2] |
| Scale-up Feasibility | Well-established for scaling [2] | Challenges in large-scale production [6] |
| Cost Considerations | Moderate to high | Low to moderate [3] |
The comparative analysis reveals distinct advantage profiles for each polymer. PLGA's synthetic nature provides precisely controllable degradation and release kinetics, making it particularly suitable for long-acting injectable formulations where predictable sustained release over weeks or months is required [4]. Its versatility is evidenced by commercial products like Lupron Depot for hormone therapy and Zilretta for osteoarthritis pain [4]. Chitosan's natural origin confers inherent bioactivity including mucoadhesion and antimicrobial properties, making it ideal for mucosal delivery systems and applications where infection control is beneficial [3]. The cationic nature of chitosan enables strong electrostatic interactions with negatively charged mucosal surfaces and biological membranes, enhancing residence time and absorption at these sites [2].
Experimental Data and Methodologies
Formulation Protocols
PLGA Nanoparticle Preparation via Emulsion-Solvent Evaporation The emulsion-solvent evaporation method is widely employed for PLGA nanoparticle synthesis. The standard protocol involves: (1) Dissolving PLGA polymer and the drug in a water-immiscible organic solvent (typically dichloromethane or ethyl acetate); (2) Emulsifying this organic phase in an aqueous solution containing a stabilizer (commonly polyvinyl alcohol - PVA) using high-speed homogenization or sonication to form an oil-in-water emulsion; (3) evaporating the organic solvent under reduced pressure with continuous stirring to solidify the nanoparticles; (4) Collecting nanoparticles by ultracentrifugation and washing to remove residual solvents and stabilizers [5]. This method enables control over particle size through parameters such as homogenization speed/sonication energy, surfactant concentration, and organic-to-aqueous phase ratio.
Chitosan Nanoparticle Preparation via Ionic Gelation Ionic gelation is the most prevalent method for chitosan nanoparticle formation, leveraging the electrostatic interaction between chitosan's cationic amino groups and polyanionic cross-linkers. The standard procedure includes: (1) Dissolving chitosan in a dilute acidic aqueous solution (typically 1% acetic acid) to protonate the amino groups; (2) Preparing a separate aqueous solution of a polyanionic cross-linker, most commonly tripolyphosphate (TPP); (3) Adding the TPP solution dropwise to the chitosan solution under constant magnetic stirring at room temperature, leading to instantaneous gelation via electrostatic cross-linking; (4) Continuing stirring to allow nanoparticle maturation [3]. Critical parameters affecting nanoparticle characteristics include the chitosan molecular weight and degree of deacetylation, chitosan-to-TPP mass ratio, pH conditions, and stirring speed.
Characterization Data and Analytical Techniques
Comprehensive characterization of polymeric nanoparticles is essential for quality control and predictive performance assessment. Key parameters include size, surface charge, morphology, drug loading efficiency, and in vitro release profile.
Table 3: Standard Characterization Techniques for Polymeric Nanoparticles
| Parameter | Analytical Technique | Protocol Details | Significance |
|---|---|---|---|
| Size Distribution | Dynamic Light Scattering (DLS) [1] | Measurement of particle Brownian motion | Affects circulation time, biodistribution, cellular uptake |
| Surface Charge | Zeta Potential Analysis [1] | Measurement of electrophoretic mobility | Predicts colloidal stability and biological interactions |
| Particle Morphology | Transmission Electron Microscopy (TEM) [1] | High-resolution imaging | Reveals nanoparticle shape and internal structure |
| Chemical Structure | Nuclear Magnetic Resonance (NMR) [1] | Analysis of polymer and drug signals | Confirms polymer composition and drug conjugation |
| Drug Loading | HPLC/UV-Vis Spectroscopy [2] | Measurement of drug content after extraction | Determines encapsulation efficiency and loading capacity |
| In Vitro Release | Dialysis Method [4] | Sampling and analysis of released drug over time | Predicts in vivo release kinetics and duration |
Recent studies have provided quantitative performance comparisons. A comprehensive analysis of PLGA microparticles encompassing 321 in vitro release studies demonstrated that formulation parameters significantly impact release characteristics, with polymer molecular weight (typically 12-75 kDa) and lactide:glycolide ratio (commonly 50:50 or 75:25) being primary determinants [4]. Research on chitosan nanoparticles has highlighted their exceptional mucoadhesive properties, with studies demonstrating up to 3-5 times improved bioavailability for drugs delivered via nasal and oral mucosal routes compared to conventional formulations [2].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagents for Polymeric Nanoparticle Research
| Reagent/Material | Function in Research | Application Examples |
|---|---|---|
| PLGA Polymer | Primary matrix for nanoparticle formation | Controlled release formulations, injectable depots [4] |
| Chitosan | Natural polymer carrier | Mucosal delivery, wound healing applications [3] |
| Polyvinyl Alcohol (PVA) | Stabilizer/surfactant in emulsion methods | Prevents aggregation during PLGA nanoparticle formation [5] |
| Tripolyphosphate (TPP) | Ionic crosslinker for chitosan | Forms nanoparticles via ionic gelation [3] |
| Dichloromethane | Organic solvent for polymer dissolution | Solvent in emulsion-solvent evaporation method [5] |
| Acetic Acid | Solubilizing agent for chitosan | Protonates amino groups for aqueous dissolution [3] |
| Polyethylene Glycol (PEG) | Surface modifier for stealth properties | PEGylation to prolong circulation time [5] |
| Coumarin-6 | Fluorescent tracer for imaging | Tracking cellular uptake and biodistribution [1] |
Research Workflow and Experimental Design
Figure 1: Experimental workflow for polymeric nanoparticle research and development.
Contextualizing Polymeric Nanoparticles vs. Liposomal Formulations
In the broader landscape of nanomedicine research, polymeric nanoparticles represent a distinct category alongside lipid-based systems, particularly liposomes. While both are versatile drug delivery platforms, they exhibit fundamental differences in composition, structure, and functional characteristics. Liposomes are spherical vesicles composed of concentric phospholipid bilayers enclosing aqueous compartments, structurally mimicking biological membranes [7] [8]. This architecture allows for simultaneous encapsulation of both hydrophilic drugs (within the aqueous core) and hydrophobic drugs (within the lipid bilayers) [7].
The historical development and clinical translation of these systems have followed different trajectories. Liposomes have a longer history of clinical use, with pioneering products like Doxil (doxorubicin-loaded liposomes) establishing the clinical viability of nanocarriers [8]. Their modular composition, typically including phospholipids and cholesterol, provides excellent biocompatibility but can present stability challenges [7]. Polymeric nanoparticles, particularly PLGA-based systems, offer superior stability and more tunable drug release profiles, with degradation kinetics that can be precisely controlled through polymer composition [2].
A significant advancement for both platforms has been the implementation of PEGylation—the surface attachment of polyethylene glycol chains—to create "stealth" nanoparticles that evade immune recognition and prolong circulation half-life [8] [5]. For liposomes, this addresses rapid clearance by the mononuclear phagocyte system, while for polymeric nanoparticles, it enhances bioavailability and target site accumulation [8] [5]. Current research in both fields is increasingly focused on active targeting strategies through surface functionalization with ligands that recognize specific receptors overexpressed on target cells [7] [5].
PLGA and chitosan nanoparticles represent two advanced platforms within the broader category of polymeric drug delivery systems, each with distinctive advantages tailored to specific therapeutic applications. PLGA's superior controlled release capabilities, tunable degradation kinetics, and extensive regulatory approval history make it ideal for sustained-release formulations, particularly for chronic conditions requiring long-term therapy [4] [2]. Chitosan's natural origin, inherent mucoadhesiveness, antimicrobial properties, and permeability-enhancing effects position it as an excellent candidate for mucosal delivery systems and localized therapies [3] [2].
The future trajectory of polymeric nanoparticle development is likely to focus on several key areas: First, the creation of hybrid systems that combine advantageous properties of multiple polymers, such as PLGA-chitosan composites that offer both controlled release and enhanced mucosal adhesion [2]. Second, the development of "smart" nanoparticles with stimuli-responsive properties that release their payload in response to specific pathological triggers such as pH, enzyme activity, or temperature [1]. Third, the integration of artificial intelligence and machine learning approaches to accelerate nanoparticle design and optimization, as demonstrated by recent efforts to create comprehensive datasets of PLGA formulation parameters [4]. Finally, increased attention to sustainable nanocarrier design principles, including green synthesis methods and environmentally benign materials, will likely shape future research directions [6].
When positioned within the comprehensive landscape of nanomedicine platforms, polymeric nanoparticles offer distinct advantages in stability and controlled release compared to lipid-based systems, while liposomes maintain benefits in encapsulation versatility and clinical track record. The continued refinement of both platforms, along with the emergence of increasingly sophisticated hybrid approaches, promises to significantly advance targeted therapeutic interventions across a broad spectrum of human diseases.
Liposomes, spherical vesicles comprising one or more phospholipid bilayers enclosing an aqueous core, stand as a cornerstone in drug delivery systems. Their architecture, mimicking biological membranes, confers exceptional biocompatibility and the unique ability to encapsulate both hydrophilic (in the aqueous core) and hydrophobic (within the lipid bilayer) therapeutic agents [9] [10]. Since their discovery, liposomes have been extensively researched and commercialized, with formulations like Doxil paving the way for targeted and sustained drug delivery [10]. Within the broader thesis of analyzing polymeric nanoparticles versus liposomal formulations, it is crucial to understand that the performance of liposomes is not merely a function of their vesicular structure but is profoundly governed by their molecular composition and the physical state of the lipid bilayer [11] [12]. This guide objectively unpacks the critical roles of phospholipid bilayers, cholesterol, and membrane fluidity in shaping the critical quality attributes (CQAs) of liposomal formulations, providing direct comparisons with polymeric nanoparticle systems where pertinent experimental data exists.
Composition and Structure: The Liposome Framework
The Phospholipid Bilayer Foundation
The primary structural component of a liposome is the phospholipid bilayer. These amphiphilic lipids spontaneously assemble into bilayers in an aqueous environment, forming a barrier that separates the internal volume from the external medium [10]. The choice of phospholipid—such as the commonly used phosphatidylcholine (PC)—is a critical formulation decision, as it influences fundamental properties like membrane rigidity, surface charge, and biocompatibility [13]. Notably, phospholipids are not biologically inert; recent studies demonstrate that liposomes composed of palmitoyl oleoyl phosphatidylcholine (POPC) can significantly modulate gene expression in macrophages, particularly upregulating inflammatory pathways via NF-κB activation [13].
Cholesterol: The Membrane Stabilizer
Cholesterol is a ubiquitous and crucial component integrated into the phospholipid bilayer. Its role extends far beyond a simple structural filler. Cholesterol modulates membrane fluidity and stability by inserting itself between the phospholipid hydrocarbon chains, restricting their motion and increasing the packing density of the bilayer [14] [13]. This action leads to a more mechanically robust membrane that is less permeable to water-soluble molecules and reduces the risk of liposome fusion or aggregation during storage [9]. Furthermore, its incorporation is biologically relevant; studies show that including free cholesterol (30%) in POPC liposomes significantly attenuates the NF-κB-mediated inflammatory response they would otherwise induce in macrophages [13]. This effect is specific to free cholesterol and is not observed with esterified or water-soluble forms, highlighting a functional role beyond mere structural support [13].
Table 1: Key Components of Liposomal Formulations and Their Functions
| Component | Type | Primary Function | Impact on Critical Quality Attributes (CQAs) |
|---|---|---|---|
| Phospholipids (e.g., POPC) | Structural Lipid | Forms the foundational bilayer structure [13] [10]. | Determines intrinsic membrane fluidity, charge, and biocompatibility. Can influence immunogenicity [13]. |
| Cholesterol | Membrane Stabilizer | Modulates fluidity and reduces permeability [14] [13]. | Enhances physical stability, reduces drug leakage, and can suppress inflammatory responses [13]. |
| PEGylated Lipids | Functional Excipient | Creates a hydrophilic "stealth" corona around the liposome [9]. | Prolongs circulation time by reducing opsonization and recognition by the immune system [9]. |
Comparative Performance: Liposomes vs. Polymeric Nanoparticles
Direct comparative studies provide valuable insights into the relative strengths and weaknesses of liposomal and polymeric nanoparticle formulations. A 2020 study directly compared L-carnitine-loaded liposomes (lipo-carnitine) with poly(lactic-co-glycolic acid) (PLGA) nanoparticles (nano-carnitine), characterizing their physicochemical properties and release profiles [11]. The data reveal a clear trade-off between particle characteristics and encapsulation efficiency.
Table 2: Experimental Comparison of L-Carnitine-loaded Liposomes vs. PLGA Nanoparticles [11]
| Formulation | Particle Size (nm) | Polydispersity Index (PDI) | Zeta Potential (mV) | Encapsulation Efficiency (%) |
|---|---|---|---|---|
| Lipo-carnitine (Liposome) | 97.88 ± 2.96 | 0.35 ± 0.01 | +6.36 ± 0.54 | 14.26 ± 3.52 |
| Nano-carnitine (PLGA) | 250.90 ± 6.15 | 0.22 ± 0.03 | -32.80 ± 2.26 | 21.93 ± 4.17 |
The data shows that the liposomal formulation achieved a smaller particle size and a near-neutral surface charge, while the polymeric PLGA nanoparticles offered superior drug encapsulation and a more monodisperse size distribution (lower PDI) [11]. Both systems successfully transformed the rapid-release profile of free L-carnitine (90% release within 1 hour) into a controlled-release profile sustained over 12 hours, demonstrating their efficacy as delivery systems [11].
Another study on nose-to-brain delivery of meloxicam found that solid lipid nanoparticles (SLNs), a subtype of lipid nanoparticles, showed higher encapsulation efficiency and drug loading than PLGA nanoparticles. Furthermore, chitosan-coated SLNs demonstrated superior in vitro release, mucoadhesion, and permeation behavior compared to both PLGA NPs and the native drug [12].
Membrane Fluidity: A Pivotal Critical Quality Attribute
Membrane fluidity describes the dynamic, lateral movement and flexibility of lipid molecules within the bilayer [14]. It is a central CQA that acts as a measurable indicator of bilayer behavior, directly impacting drug release kinetics, stability, and biological interactions [14]. Fluidity should not be confused with the phase transition temperature (Tm); rather, it is a reflection of the membrane's physical state at a given temperature.
Experimental Assessment of Fluidity
Fluidity is most practically assessed using fluorescence-based probes, which offer high sensitivity and are amenable to high-throughput screening [14]. Common probes include:
- DPH (1,6-diphenyl-1,3,5-hexatriene): A hydrophobic probe that aligns with the fatty acid chains, reporting on the order of the acyl chain region.
- Laurdan (6-lauroyl-2-dimethylaminonaphthalene): Sensitive to the polarity of its environment, it can distinguish between gel (ordered) and liquid crystalline (disordered) phases through Generalized Polarization (GP) measurements [14].
Factors Governing Membrane Fluidity
Membrane fluidity is influenced by a complex, interdependent set of factors, which form the basis of rational formulation design [14]:
- Cholesterol Content: Increasing cholesterol concentration up to ~30-50% generally reduces fluidity by restricting phospholipid chain motion, leading to a more condensed and stable membrane [14] [13].
- Lipid Acyl Chain Length and Saturation: Shorter chains and a higher degree of unsaturation (double bonds) in fatty acids increase fluidity by reducing intermolecular interactions and introducing kinks that prevent tight packing.
- Temperature: Higher temperatures increase the kinetic energy of lipids, thereby increasing membrane fluidity.
- PEGylated Lipids: The incorporation of PEG-lipids can increase the viscosity of the membrane surface and influence fluidity, albeit in a complex manner that depends on the PEG chain length and density [14].
Diagram 1: Factors influencing membrane fluidity and its impact on performance.
Advanced Applications and Synergistic Systems
The understanding of liposomal components has enabled the development of advanced, hybrid nanomedical applications. A prominent example is the synergy between gold nanoparticles (AuNPs) and lipid membranes. Functionalizing AuNPs with lipid membranes significantly enhances their biocompatibility and stability in biological environments, reducing cytotoxicity and mitigating rapid clearance by the immune system [15]. Furthermore, this combination unlocks theranostic (therapeutic + diagnostic) capabilities. Membrane-embedded AuNPs can act as nanoscale heaters, enabling spatiotemporally controlled drug release through light-triggered lipid phase transitions, a property not achievable by either component alone [15].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Liposome Research and Development
| Reagent / Material | Function in Formulation | Experimental Application / Notes |
|---|---|---|
| Phosphatidylcholines (e.g., POPC) | Primary phospholipid for bilayer formation [13]. | Provides a biocompatible, zwitterionic backbone. A common choice for model membranes. |
| Cholesterol | Membrane stabilizer and fluidity modulator [14] [13]. | Use free cholesterol, not esterified forms, for incorporation into the bilayer for optimal effect on membrane properties and inflammatory response suppression [13]. |
| PEGylated Phospholipids (e.g., DSPE-PEG) | Confers "stealth" properties to prolong circulation half-life [9]. | Critical for reducing protein adsorption and rapid clearance. A key component in mRNA COVID-19 vaccines [9]. |
| Fluorescent Probes (e.g., DPH, Laurdan) | Molecular sensors for biophysical characterization [14]. | Essential for experimentally quantifying membrane fluidity and phase behavior via fluorescence spectroscopy. |
| Cationic Lipids | Enables encapsulation of nucleic acids (DNA, mRNA) via charge interaction. | Core component of lipid nanoparticles (LNPs) for gene delivery and vaccines [9]. |
Experimental Protocols for Key Investigations
Protocol: Assessing the Anti-inflammatory Effect of Cholesterol
This protocol is derived from experiments demonstrating cholesterol's ability to attenuate liposome-induced inflammation [13].
Liposome Preparation:
- Prepare two liposome batches using a mini-extruder: one with 100% POPC and another with a 70:30 molar ratio of POPC to free cholesterol.
- Dry the lipid mixtures from chloroform under a nitrogen stream and hydrate the film in endotoxin-free Tris buffer (pH 7.4).
- Extrude the suspension 15 times through a 100 nm membrane filter.
- Purify via ultracentrifugation (100,000 × g, 60 min, 4°C) and resuspend in sterile PBS.
- Characterize size and concentration using Nanoparticle Tracking Analysis (NTA) and ensure endotoxin levels are below 0.01 EU/mL.
Cell Treatment and Analysis:
- Culture macrophage cell lines (e.g., RAW264.7 or J774A.1).
- Incubate cells with equal amounts (e.g., based on lipid mass) of POPC and POPC-Cholesterol liposomes for a set period (e.g., 6 hours).
- Quantify the inflammatory response by measuring TNF-α expression levels using RT-qPCR or ELISA.
- Investigate the mechanism by analyzing NF-κB activation, for instance, via Western blot for phosphorylated NF-κB subunits.
Protocol: Investigating Drug Release Kinetics
This protocol outlines a general approach for conducting in vitro release (IVR) studies, a critical CQA test [16].
Formulation Screening:
- Prepare a series of liposomal formulations varying key parameters (e.g., cholesterol content from 0% to 40%, different phospholipid saturations).
IVR Test Setup:
- Use a standardized apparatus such as a dialysis system or sample-and-separate method.
- Select appropriate release media (e.g., PBS at pH 7.4) and maintain at a constant physiological temperature (37°C) with continuous stirring.
- Place the liposome formulation in a dialysis bag or directly into the release medium.
Sampling and Analysis:
- At predetermined time intervals, withdraw aliquots of the release medium and replace with fresh medium to maintain sink conditions.
- Analyze the drug concentration in the samples using a validated analytical method (e.g., HPLC, UV-Vis spectroscopy).
- Calculate the cumulative percentage of drug released over time.
Data Modeling:
- Fit the obtained release data to kinetic models (e.g., Weibull, Higuchi, zero-order) to understand the release mechanism.
- Use machine learning workflows to correlate formulation parameters with the resulting release profiles for accelerated development [16].
Diagram 2: Integrated workflow for liposomal formulation characterization.
The performance of liposomal formulations is intricately linked to their nanoscale architecture and composition. The phospholipid bilayer provides the foundational structure, while cholesterol serves as a critical modulator of membrane fluidity, stability, and even biological interactions. As demonstrated by direct comparative studies, liposomes often provide distinct advantages in terms of size and surface charge compared to polymeric nanoparticles like PLGA, which may offer superior encapsulation efficiency and monodispersity [11] [12]. A deep understanding of membrane fluidity as a pivotal CQA enables formulators to rationally design liposomes with optimized drug release profiles, stability, and therapeutic efficacy. The ongoing integration of advanced techniques, including machine learning for predicting release profiles [16] and the development of hybrid systems like AuNP-lipid composites [15], continues to push the boundaries of liposomal technology in nanomedicine.
In the field of nanomedicine and drug delivery, the structural architecture of carrier systems is a critical determinant of their performance. This guide provides an objective comparison of two fundamental structural classifications: unilamellar versus multilamellar vesicles in liposomal systems, and solid versus core-shell matrices in polymeric nanoparticles. Understanding these distinctions is essential for researchers and drug development professionals to select the optimal nanocarrier for specific therapeutic applications, balancing factors such as drug loading, release kinetics, stability, and biological interactions. The systematic analysis presented here, supported by experimental data and protocols, aims to inform rational design choices in pharmaceutical development.
Vesicle Architectures: Unilamellar vs. Multilamellar
Liposomal vesicles are primarily classified based on their lamellar structure, which significantly influences their drug encapsulation capacity, release profile, and biological activity.
Structural Characteristics and Experimental Performance
Table 1: Structural and Functional Comparison of Vesicle Architectures
| Parameter | Unilamellar Vesicles (ULV) | Multilamellar Vesicles (MLV) |
|---|---|---|
| Structural Definition | Single phospholipid bilayer surrounding an aqueous core [10] | Multiple concentric phospholipid bilayers separated by aqueous compartments [10] |
| Typical Size Range | 20 nm to >1 μm [10] | Typically larger, often exceeding 1 μm [10] |
| Drug Encapsulation | Hydrophilic drugs in aqueous core; hydrophobic drugs in lipid bilayer [10] | Higher capacity for both hydrophilic and hydrophobic drugs due to multiple compartments |
| Antibody Response (PFC) | Vigorous; Significantly higher than MLV-BSA [17] | Vigorous; lower than ULV-BSA [17] |
| Key Advantage | More efficient contact with immune cells due to simpler structure [17] | Higher encapsulation capacity for some compounds |
Experimental Evidence and Protocol
A seminal study directly compared the effectiveness of Multilamellar Vesicles (MLV) and Unilamellar Vesicles (ULV) in enhancing specific antibody formation [17].
- Vesicle Preparation: MLV and ULV were composed of dimyristoyl-lecithin, cholesterol, and dicetyl phosphate in a molar ratio of 7:2:1. Bovine Serum Albumin (BSA) was used as the model antigen, and liposome-associated BSA was purified via Blue Sepharose CL-6B column chromatography [17].
- Structural Confirmation: Electron microscopy was used to confirm the presence of appropriate lamellar structures for each preparation [17].
- In Vivo Evaluation: Mice were injected with either free BSA, empty vesicles, or liposome-associated BSA (MLV-BSA or ULV-BSA). The plaque-forming cell (PFC) response was measured to quantify the specific antibody formation [17].
- Key Finding: While both liposome-associated BSA formulations generated a vigorous PFC response, the magnitude of the response induced by BSA entrapped in unilamellar vesicles was significantly higher than that in multilamellar vesicles. This suggests the lamellar arrangement plays a crucial role in affecting the potentiated antibody response, potentially due to more efficient cellular processing of the simpler ULV structure [17].
Experimental Workflow for Vesicle Comparison
Polymeric Matrices: Solid vs. Core-Shell
Polymeric nanoparticles offer an alternative to lipid-based systems, with their own structural classifications that govern functionality. The primary distinction lies between solid matrix systems and core-shell architectures.
Structural Definitions and Comparative Performance
Table 2: Structural and Functional Comparison of Polymeric Nanoparticles
| Parameter | Solid Matrix Systems (SLNs) | Nanostructured Lipid Carriers (NLCs) | Core-Shell Polymeric Hybrids (PLNs) |
|---|---|---|---|
| Structural Definition | Solid lipid matrix at room temperature [18] [10] | Mixed solid and liquid lipid matrix, creating an imperfect structure [18] [10] | Polymeric core surrounded by a lipid/PEG shell [19] |
| Drug Loading Capacity | ~80% for HCT [18] | ~90% for HCT [18] | High, due to combined polymer-lipid structure [19] |
| Drug Release (300 min) | ~65% of HCT released [18] | >90% of HCT released [18] | Controlled release from polymeric core [19] |
| Storage Stability (6 mo) | ~15% drug loss [18] | <5% drug loss [18] | High physical and storage stability [19] |
| Key Advantage | Biocompatibility, solid matrix protection [18] [10] | Superior drug loading and stability vs. SLNs [18] | Combines stability of polymers with biocompatibility of lipids [19] |
Core-Shell Structures: Solid vs. Hollow
A further distinction within core-shell architectures is the nature of the core itself. Solid Core-Shell (SCs-MIP) structures have a solid interior, while Hollow Core-Shell (HCs-MIP) structures possess an empty interior space [20].
- Diffusion Performance: Research on molecularly imprinted polymers has demonstrated that the diffusion coefficient (D) of probe molecules in HCs-MIP is approximately 1.5 times higher than in SCs-MIP. This enhancement is attributed to the bilateral mass diffusion of analyte or probe molecules from both the outer and inner interfaces of the MIP layer [20].
- Analytical Application: This superior diffusion profile makes hollow core-shell structures particularly advantageous for applications requiring high sensitivity, such as electrochemical sensing of anti-HIV drugs like Lamivudine (3TC) and Zidovudine (AZT) [20].
Diffusion Pathways in Core-Shell Structures
Experimental Protocol for Lipid Nanoparticle Formulation
The following methodology details the preparation and optimization of solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs), as used in a study on the diuretic drug Hydrochlorothiazide (HCT) for pediatric therapy [18].
1. Material Screening:
- Solubility Studies: The solubilizing power of various solid lipids (e.g., Precirol ATO5, Compritol 888 ATO) and liquid lipids (e.g., Transcutol HP, Miglyol 812) towards HCT is assessed. For solid lipids, drug solubility is visually estimated in the melted lipid. For liquid lipids, equilibrium solubility is determined after 24 hours at 65°C [18].
- Compatibility Studies: Differential Scanning Calorimetry (DSC) is used to analyze the compatibility between the drug and formulation components by scanning physical mixtures under static air [18].
2. Nanoparticle Preparation:
- The Hot High-Shear Homogenization (HSH) technique is employed.
- The lipid phase (solid lipid alone for SLNs, or a blend of solid and liquid lipids for NLCs) is heated to 65°C to melt.
- An aqueous surfactant phase (containing surfactants like Gelucire 44/14, Tween 80, or Pluronic F68) is heated to the same temperature.
- The aqueous phase is dispersed into the lipid phase using high-shear homogenization (e.g., at 10,000 rpm for 5-10 minutes) to form a pre-emulsion.
- The resulting pre-emulsion is further processed by ultrasonication to reduce particle size and achieve a uniform dispersion [18].
3. Characterization:
- Particle Size & PDI: Dynamic light scattering measures mean particle diameter and polydispersity index (PDI).
- Zeta Potential: Laser Doppler electrophoresis assesses surface charge.
- Encapsulation Efficiency (EE%): Determined by quantifying the amount of successfully entrapped drug versus the initial total amount.
- In Vitro Release Study: The drug release profile is evaluated using dialysis membranes in a suitable release medium under sink conditions [18].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Nanoparticle Formulation and Evaluation
| Reagent Category | Specific Examples | Function in Research |
|---|---|---|
| Solid Lipids | Precirol ATO5, Compritol 888 ATO, Glyceryl Monostearate (Geleol) [18] | Forms the solid matrix of SLNs and the solid part of NLCs; determines drug loading and release kinetics. |
| Liquid Lipids | Transcutol HP, Caprylic/Capric Triglycerides (Miglyol, Labrafac) [18] | Creates imperfections in the solid lipid matrix of NLCs to improve drug loading and prevent expulsion. |
| Surfactants | Gelucire 44/14, Tween 80, Pluronic F68, Poloxamer 188 [18] | Stabilizes the nanoparticle dispersion during and after formation; critical for controlling particle size and stability. |
| Phospholipids | DSPC, DOPE, HSPC [21] [19] | Primary component of liposomal bilayers and the lipid shell in hybrid systems; provides biocompatibility. |
| Biodegradable Polymers | PLGA, PLGA-mPEG [11] [19] | Forms the core of polymeric and hybrid nanoparticles; degrades in the body to provide controlled drug release. |
| PEGylated Lipids | DMG-PEG, DSG-PEG, ALC-0159 [21] [19] | Creates a "stealth" coating on nanoparticles to reduce immune clearance and prolong circulation half-life. |
| Cationic Lipids | DLin-MC3-DMA, ALC-0315, SM-102 [21] | Enables encapsulation of nucleic acids (mRNA, siRNA) in lipid nanoparticles via electrostatic interaction. |
The choice between unilamellar and multilamellar vesicles or solid and core-shell polymeric matrices is not a matter of superiority but of application-specific optimization. Unilamellar vesicles demonstrate advantages in scenarios requiring efficient biological interaction, such as adjuvant activity, while multilamellar vesicles offer higher encapsulation capacity. In the polymeric domain, solid lipid nanoparticles provide a robust and biocompatible platform, but nanostructured lipid carriers and advanced core-shell systems like polymeric lipid hybrid nanoparticles (PLNs) address limitations in drug loading and stability. The emerging class of hollow core-shell particles further extends functionality by enhancing mass transport. The experimental data and protocols summarized in this guide provide a foundation for researchers to make informed decisions in the design and development of next-generation nanomedicines.
In the evolving landscape of nanomedicine, polymeric nanoparticles (PNPs) and liposomal formulations have emerged as frontrunner drug delivery systems. Both platforms are designed to enhance the therapeutic efficacy of active pharmaceutical ingredients, yet they are distinguished by their unique structural compositions, core characteristics, and subsequent performance in biological environments. Polymeric nanoparticles are typically solid colloidal particles fabricated from natural or synthetic polymers, whereas liposomes are spherical vesicles comprising one or more phospholipid bilayers surrounding an aqueous core [22] [23]. This fundamental architectural difference dictates their interactions with biological systems, their capacity to encapsulate diverse therapeutic agents, and their inherent stability profiles.
The selection of an appropriate nanocarrier is a critical determinant in the success of a drug product. For researchers and drug development professionals, a clear, data-driven understanding of the inherent advantages and limitations of each system is indispensable for matching the carrier to the therapeutic application. This guide provides a comparative analysis of PNPs and liposomes, focusing on the three pivotal axes of biocompatibility, encapsulation capacity, and initial stability, supported by experimental data and standardized protocols to inform early-stage formulation development.
Comparative Analysis: Core Characteristics at a Glance
The table below summarizes the fundamental properties of polymeric nanoparticles and liposomes, providing a high-level overview of their key characteristics.
Table 1: Inherent Properties of Polymeric Nanoparticles and Liposomal Formulations
| Characteristic | Polymeric Nanoparticles (PNPs) | Liposomal Formulations |
|---|---|---|
| Core Composition | Solid polymer matrix (e.g., PLGA, Chitosan) [24] [25] | Phospholipid bilayer(s) enclosing an aqueous core [23] [26] |
| Structural Versatility | Nanospheres, nanocapsules, polymeric micelles [24] | Unilamellar, Oligolamellar, Multilamellar Vesicles [26] |
| Biocompatibility | Ranges from excellent (natural polymers) to tunable (synthetic polymers); degradation byproducts may require evaluation [24] [25] | Generally excellent due to biomimetic lipid composition; high biological acceptance [23] [26] |
| Encapsulation Drive | Hydrophobic interactions, polymer-drug affinity, and physical entrapment within a solid matrix [22] [1] | Partitioning based on drug solubility: hydrophobic (bilayer) vs. hydrophilic (aqueous core) [23] [8] |
| Typical Drug Loading Capacity | Can be very high, particularly for hydrophobic drugs within the polymer matrix [24] | Moderate; constrained by bilayer volume/area for lipophilic drugs and core volume for hydrophilic drugs [23] |
| Inherent Physical Stability | Generally high; solid matrix minimizes drug leakage and offers robust colloidal stability [1] [27] | Can be lower; susceptible to drug leakage, fusion, and aggregation over time without formulation optimization [23] [28] |
Deep Dive into Key Performance Parameters
Biocompatibility and Biological Interaction
Polymeric Nanoparticles (PNPs): The biocompatibility of PNPs is intrinsically linked to the polymer choice. Natural polymers like chitosan and gelatin are prized for their excellent biocompatibility, biodegradability, and low toxicity [25]. Synthetic polymers such as PLGA (poly(lactic-co-glycolic acid)) are widely used because they are biodegradable and biocompatible, with degradation products (lactic and glycolic acids) that are metabolized via normal physiological pathways [24] [27]. However, the potential for cytotoxicity or immune responses must be evaluated on a case-by-case basis, particularly for novel synthetic polymers or specific degradation byproducts [22].
Liposomal Formulations: Liposomes exhibit superior biocompatibility from the outset, as they are typically composed of phospholipids and cholesterol, molecules that are natural components of biological membranes [23] [26]. This biomimetic nature makes them well-tolerated, minimally immunogenic, and biodegradable. Their surface charge can be tailored (cationic, anionic, or neutral) to influence cellular interactions, with cationic liposomes sometimes showing increased cytotoxicity compared to their anionic or neutral counterparts [26].
Encapsulation Capacity and Loading Strategies
Polymeric Nanoparticles (PNPs): PNPs offer significant versatility in encapsulation. Their solid matrix is highly effective at encapsulating hydrophobic drugs with high loading capacity, protecting them from the aqueous environment [24]. A key advantage is the ability to achieve controlled and sustained drug release profiles based on polymer erosion and diffusion mechanisms [22]. Furthermore, PNPs can be engineered as "smart" systems that respond to specific stimuli (e.g., pH, temperature, enzymes) for precise drug release at the target site [22] [1]. For instance, a study on pH-responsive PLGA NPs demonstrated a drug loading capacity of 1.8 wt% for a modified doxorubicin, leveraging the hydrophobic core for efficient encapsulation [27].
Liposomal Formulations: Liposomes possess a unique amphiphilic structure that allows for the simultaneous encapsulation of both hydrophilic drugs (within the aqueous core) and hydrophobic drugs (within the lipid bilayer) [23] [8]. However, their loading capacity is constrained by the finite volumes of these compartments. A primary strategy to enhance loading, particularly for hydrophilic drugs, is active (remote) loading, which uses transmembrane ion or pH gradients to trap ionizable drugs inside the liposome at high concentrations [23]. Experimental data for sesquiterpene lactones encapsulated in liposomes demonstrated encapsulation efficiencies of approximately 70-80%, highlighting the effectiveness of passive incorporation for lipophilic molecules [28].
Table 2: Experimental Encapsulation and Stability Data from Case Studies
| Formulation Detail | Polymeric Nanoparticle (ATRAM-BSA-PLGA) [27] | Liposomal Formulation (Soybean PC) [28] |
|---|---|---|
| Encapsulated Agent | Doxorubicin-TPP (hydrophobic) | Eremantholide C / Goyazensolide (lipophilic) |
| Reported Loading/EE | Loading Capacity: 1.8 wt% | Encapsulation Efficiency: ~70-80% |
| Stability Conditions | In cell culture medium with 10% FBS for 72 hours | Storage at 4°C over 12 months; simulated GI fluids |
| Key Stability Finding | Hydrodynamic diameter remained stable with only a modest ~30 nm increase in 50% serum. | Substance concentration remained stable over 12 months; stable in simulated gastric & intestinal fluids. |
| Implied Advantage | High stability in biologically relevant media; minimal drug leakage. | Good long-term storage stability and resilience to physiological pH environments. |
Initial Stability Profile
Polymeric Nanoparticles (PNPs): PNPs generally exhibit superior physical stability. Their solid polymeric core is highly effective at preventing the rapid leakage of encapsulated drugs, a challenge often faced by other nanocarriers [1]. The covalent cross-linking in some PNP designs, such as the BSA-wrapped PLGA NPs, further enhances encapsulation stability and prevents premature drug release, even in the presence of serum proteins [27].
Liposomal Formulations: The stability of liposomes is a well-documented challenge. They can be prone to physical instability, including aggregation, fusion, and drug leakage, which can be triggered by temperature variations, mechanical stress, or interaction with biological fluids [23]. Chemically, phospholipids are susceptible to hydrolysis and oxidation, which can compromise the integrity of the bilayer [23]. Consequently, liposomal formulations often require sophisticated optimization, such as lyophilization (freeze-drying) with cryoprotectants for long-term storage, and surface modifications like PEGylation to improve stability in circulation [23] [8].
Essential Experimental Protocols for Characterization
To generate reproducible and comparable data on these nanocarriers, researchers must adhere to standardized characterization protocols. Below are detailed methodologies for two fundamental assays.
Protocol for Determining Encapsulation Efficiency (EE) and Drug Loading (DL)
This protocol is adapted from procedures used in the cited research for both PNPs and liposomes [27] [28].
- Separation of Unencapsulated Drug: Separate the nanocarriers (PNPs or liposomes) from the free, unencapsulated drug. This is typically achieved using dialysis, ultracentrifugation, or size-exclusion chromatography.
- Destruction of Nanocarriers and Drug Extraction: Lyse the separated nanocarrier pellet or suspension to release the encapsulated drug. Methods include:
- Solvent Disruption: Add an organic solvent (e.g., acetonitrile, methanol, or isopropanol) appropriate for the drug and carrier to dissolve the lipid bilayer (liposomes) or polymer matrix (PNPs). Vortex vigorously and/or sonicate to ensure complete disruption.
- Detergent Lysis: For liposomes, use a detergent like Triton X-100 to dissolve the lipid membrane.
- Quantification of Encapsulated Drug: Analyze the resulting solution using a calibrated analytical technique, such as High-Performance Liquid Chromatography (HPLC) or UV-Vis Spectrophotometry, to determine the concentration of the encapsulated drug (C_encapsulated).
- Quantification of Total Drug: Directly analyze a separate, non-purified sample of the formulation (often after dissolution/lysis as in Step 2) to determine the total drug concentration (C_total).
- Calculation:
- Encapsulation Efficiency (EE%) = (Cencapsulated / Ctotal) × 100%
- Drug Loading (DL%) = (Mass of encapsulated drug / Total mass of the nanoparticle formulation) × 100%
Protocol for Assessing In Vitro Colloidal Stability
This assay evaluates the nanoparticle's ability to maintain its size and polydispersity under storage or physiological conditions, a key indicator of aggregation and physical stability [27] [28].
- Sample Preparation: Dilute the nanocarrier formulation (PNP or liposome) in the desired medium to a suitable concentration for dynamic light scattering (DLS) measurement. Relevant media include:
- Storage buffer (e.g., PBS at 4°C, 25°C, 37°C).
- Biologically relevant fluids (e.g., phosphate-buffered saline (PBS), cell culture medium, simulated gastric/intestinal fluid).
- Incubation: Incubate the samples under controlled conditions (temperature, agitation) for a predetermined period (e.g., 24 hours, 72 hours, 1 month).
- Dynamic Light Scattering (DLS) Measurement: At designated time points, analyze each sample using a DLS instrument (Zetasizer).
- Measure the hydrodynamic diameter (nm).
- Measure the polydispersity index (PDI), which indicates the breadth of the size distribution. A PDI < 0.2 is generally considered monodisperse.
- Measure the zeta potential (mV) in a suitable electrolyte to assess surface charge stability.
- Data Analysis: Plot the mean diameter and PDI over time. A stable formulation will show minimal change in size and a consistently low PDI. A significant increase in diameter and/or PDI indicates aggregation and poor colloidal stability.
Visualizing Characterization Workflows
The following diagram illustrates the logical sequence and decision points in the characterization workflow for evaluating nanocarrier stability and encapsulation.
Diagram 1: A generalized experimental workflow for the initial characterization of nanocarriers, highlighting the parallel assessment of encapsulation parameters and physical stability.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents and Materials for Formulation and Characterization
| Reagent/Material | Function/Purpose | Relevant Formulation |
|---|---|---|
| PLGA | A biodegradable synthetic polymer forming the core matrix of PNPs; allows for controlled drug release. | Polymeric Nanoparticles [27] |
| Chitosan | A natural polysaccharide polymer; confers biocompatibility, mucoadhesion, and permeation enhancement. | Polymeric Nanoparticles [25] |
| Soybean Phosphatidylcholine (SPC) | A natural phospholipid mixture; primary building block for forming the lipid bilayer of liposomes. | Liposomes [28] |
| Cholesterol | Incorporated into lipid bilayers; modulates membrane fluidity and stability, reducing drug leakage. | Liposomes [23] [26] |
| Polyethylene Glycol (PEG)-Lipid | Used for surface PEGylation; confers "stealth" properties by reducing opsonization and extending circulation half-life. | Liposomes (Stealth) [8] |
| Dialysis Tubing / Filters | For purifying formulated nanocarriers by separating them from unencapsulated free drug. | Both [28] |
| Tripolyphosphate (TPP) | A polyanionic cross-linking agent used in ionic gelation to form chitosan nanoparticles. | Polymeric Nanoparticles [25] |
| Dynamic Light Scattering (DLS) Instrument | For critical quality attribute measurement: hydrodynamic particle size, PDI, and zeta potential. | Both [1] [28] |
Both polymeric nanoparticles and liposomal formulations offer distinct and powerful tools for advanced drug delivery. The choice between them is not a matter of superiority, but of strategic alignment with the therapeutic agent and the target product profile. Polymeric nanoparticles excel with high loading capacity for hydrophobic drugs, robust physical stability, and tunable, stimuli-responsive release kinetics. Liposomal formulations offer unparalleled biocompatibility, the unique ability to co-deliver hydrophilic and hydrophobic agents, and a well-established pathway for clinical translation, albeit with greater inherent challenges regarding physical and chemical stability. A deep understanding of these inherent advantages and limitations, coupled with rigorous characterization using standardized protocols, provides the foundational knowledge required to navigate the complex landscape of nanocarrier selection and optimization.
Synthesis, Engineering, and Therapeutic Deployment in Disease Targeting
Polymeric nanoparticles (PNPs) represent a cornerstone of modern nanomedicine, enabling advancements in drug delivery, bioavailability, and targeted therapy. Their clinical translation hinges on the selection and optimization of fabrication techniques that dictate critical quality attributes such as particle size, polydispersity, drug loading, and encapsulation efficiency. This guide provides a systematic comparison of three principal fabrication methods—solvent evaporation, nanoprecipitation, and microfluidics—focusing on their operational principles, resultant nanoparticle characteristics, and experimental protocols. Within the broader context of polymeric nanoparticles versus liposomal formulations research, understanding these techniques is critical for rational design of drug delivery systems. The reproducibility and scalability offered by microfluidic systems present a significant advantage over conventional bulk methods, directly addressing batch-to-batch variability challenges that have long hindered nanomedicine translation [29].
Technical Comparison of Fabrication Methods
Table 1: Comparative analysis of key fabrication techniques for polymeric nanoparticles.
| Fabrication Method | Key Mechanism | Typical Particle Size (nm) | Polydispersity Index (PDI) | Drug Loading Capacity | Encapsulation Efficiency | Scalability | Key Advantages | Major Limitations |
|---|---|---|---|---|---|---|---|---|
| Solvent Evaporation [30] | Emulsification of polymer solution into aqueous phase followed by solvent removal | 150 – 300 | Broad (High) | Moderate | Moderate for hydrophilic drugs (double emulsion) | Moderate (batch process) | Suitable for hydrophilic drugs (W/O/W) | High polydispersity, batch-to-batch variability |
| Nanoprecipitation (Batch) [31] [32] | Solvent displacement and polymer precipitation upon mixing with anti-solvent | 100 – 250 | Moderate to Broad | Moderate | Low to Moderate | Moderate (batch process) | Simplicity, no need for high-energy mixing | Low encapsulation efficiency for hydrophilic drugs |
| Microfluidics [29] [31] [30] | Hydrodynamic flow focusing for controlled mixing and nanoprecipitation | 50 – 200 (tunable) | Narrow (Low, <0.1) | High | High (~90%) [30] | High (continuous process) | Superior reproducibility, precise size control, continuous production | Low initial production rate, requires specialized equipment |
Detailed Experimental Protocols
Solvent Evaporation (Double Emulsion Method)
The double emulsion (water-in-oil-in-water, W/O/W) solvent evaporation technique is particularly suited for encapsulating hydrophilic drugs [30].
- Primary Emulsion Formation: An aqueous solution containing the hydrophilic active (e.g., a peptide or protein) is added to an organic phase containing the dissolved polymer (e.g., PLGA in dichloromethane, DCM). This mixture is subjected to high-speed homogenization or probe sonication to form a stable primary water-in-oil (W/O) emulsion.
- Secondary Emulsion Formation: The primary W/O emulsion is immediately transferred into a large volume of an external aqueous phase containing an emulsifier (e.g., polyvinyl alcohol, PVA). This secondary emulsification is typically performed using a high-speed mechanical stirrer to form a double (W/O/W) emulsion.
- Solvent Evaporation & Hardening: The double emulsion is stirred for several hours to allow the organic solvent to evaporate, solidifying the polymer and forming solid nanoparticles. The hardened nanoparticles are then collected by ultracentrifugation and washed repeatedly with purified water to remove residual solvents and emulsifiers.
Batch Nanoprecipitation
Batch nanoprecipitation relies on the spontaneous formation of nanoparticles upon the displacement of a water-miscible solvent from a polymer solution [32].
- Organic Phase Preparation: The polymer (e.g., PLGA or PLA) and the hydrophobic drug are co-dissolved in a water-miscible organic solvent such as acetone or acetonitrile. This constitutes the organic phase.
- Anti-Solvent Mixing: The organic phase is introduced, typically via syringe pump or dropwise addition under moderate magnetic stirring, into a larger volume of an anti-solvent (water, often containing a stabilizer like Poloxamer 188 or polysorbate 80).
- Nanoparticle Self-Assembly: Upon mixing, the organic solvent rapidly diffuses into the aqueous phase, reducing the solvent quality for the polymer. This leads to supersaturation, nucleation, and the spontaneous formation of solid nanoparticles as the polymer and drug precipitate out.
- Solvent Removal & Purification: The residual organic solvent is removed from the nanoparticle suspension under reduced pressure or by dialysis. The final PNPs are obtained through filtration or centrifugation.
Microfluidic-Assisted Nanoprecipitation
Microfluidic fabrication uses chip-based devices to achieve highly controlled and reproducible mixing, overcoming the limitations of bulk methods [31] [30].
- Device Setup: A common configuration is a glass or PDMS cross-shaped microfluidic chip. Syringe pumps are used to precisely control the flow rates of the fluid streams.
- Hydrodynamic Flow Focusing: The organic phase (polymer and drug in a water-miscible solvent) is injected through the central inlet. The aqueous phase (water with surfactant) is introduced through the two side inlets at a significantly higher flow rate. This geometry "focuses" the organic stream into a narrow thread, enabling rapid and uniform diffusion of the solvent into the anti-solvent.
- Continuous Nanoparticle Formation: The controlled mixing triggers nanoprecipitation in a confined region of the chip, producing nanoparticles with narrow size distribution. The nanoparticles are collected in a reservoir at the outlet in a continuous manner.
- Key Parameter Control: The Flow Rate Ratio (FRR), defined as the aqueous phase flow rate divided by the organic phase flow rate, is a critical parameter. A higher FRR typically results in smaller nanoparticle sizes due to faster mixing [31].
Table 2: Essential research reagents and materials for PNP fabrication.
| Reagent/Material | Function/Role | Example Uses & Notes |
|---|---|---|
| PLGA (Poly(lactic-co-glycolic acid)) | Biodegradable polymer matrix | Most common polymer; FDA-approved; lactide:glycolide ratio tunes degradation rate [30]. |
| PVA (Polyvinyl Alcohol) | Surfactant/Stabilizer | Used in solvent evaporation to prevent coalescence of emulsion droplets [31]. |
| Poloxamer 188 (Pluronic F68) | Surfactant/Stealth agent | Used in nanoprecipitation and microfluidics; provides steric stabilization and can impart "stealth" properties [31]. |
| Vitamin E TPGS (d-α-tocopheryl PEG succinate) | Surfactant/Stabilizer | Effective stabilizer for nanoprecipitation; can enhance drug encapsulation and efficacy [31]. |
| Acetone | Water-miscible solvent | Common solvent for nanoprecipitation and microfluidics due to its rapid diffusion into water [31] [32]. |
| DCM (Dichloromethane) | Water-immiscible solvent | Standard solvent for single and double emulsion (solvent evaporation) methods [30]. |
| Syringe Pumps | Fluid driving system | Essential for microfluidic setups and controlled batch nanoprecipitation to ensure precise, pulsation-free flow rates. |
Data, Workflows, and Visualization
Comparative Performance Data
Experimental data consistently demonstrates the superiority of microfluidics in producing monodisperse PNPs. A direct comparison of PLGA nanoparticles fabricated via bulk mixing versus microfluidic nanoprecipitation showed a significant reduction in Polydispersity Index (PDI) from over 0.2 in bulk to below 0.1 using a microfluidic chip [31]. Furthermore, microfluidic encapsulation of drugs like Doxorubicin and Paclitaxel has been reported to achieve encapsulation efficiencies exceeding 90%, a substantial improvement over many batch processes [30].
Microfluidic Nanoprecipitation Workflow
The following diagram illustrates the controlled workflow and mechanism of nanoparticle formation in a microfluidic flow-focusing device.
Size Distribution Comparison
This diagram provides a conceptual comparison of the particle size distributions characteristic of each fabrication method.
The choice of fabrication technique for polymeric nanoparticles is a critical determinant of their performance and translational potential. While solvent evaporation and batch nanoprecipitation remain valuable for specific applications, microfluidic-assisted nanoprecipitation stands out for its unparalleled control over particle characteristics, high reproducibility, and continuous operation mode. The ability of microfluidics to produce monodisperse populations of PNPs with high encapsulation efficiency directly addresses the key challenges of batch-to-batch variability and inefficient drug loading that plague conventional methods. For researchers engaged in the comparative analysis of polymeric versus liposomal nanocarriers, the precision and data-rich nature of microfluidic fabrication provide a robust platform for generating reliable and clinically relevant formulations. Future advancements will likely focus on scaling out microfluidic production through device parallelization and integrating smart, data-driven optimization to fully realize the promise of polymeric nanomedicines [29] [32].
In the evolving field of nanomedicine, the development of effective drug delivery systems is paramount for enhancing therapeutic outcomes. Among the various nanocarriers, liposomes—spherical vesicles composed of phospholipid bilayers—stand out for their biocompatibility, ability to encapsulate diverse cargo, and well-established clinical use [33] [34]. Their exploration is often framed within a broader thesis that compares them with other platforms, notably polymeric nanoparticles (PNPs). While PNPs offer superior stability and highly tunable polymer chemistry for controlled release [1], liposomes possess the distinct advantage of a biomimetic structure that closely resembles cell membranes, facilitating fusion and cellular uptake [8]. This guide provides an objective comparison of three fundamental liposome preparation techniques: Thin-Film Hydration, Ethanol Injection, and Reverse-Phase Evaporation. The selection of a preparation method is a critical first step in nanocarrier design, as it directly influences key performance parameters such as particle size, encapsulation efficiency, and suitability for scale-up, thereby dictating the vehicle's potential for success in pre-clinical and clinical applications [33] [35].
Experimental Protocols for Liposome Preparation
Thin-Film Hydration (Bangham Method)
The Thin-Film Hydration method is the most classical and widely used technique for forming multilamellar vesicles (MLVs) [36] [37].
- Step 1: Lipid Dissolution. Dissolve 2–20 mg of lipids (e.g., DPPC or POPC) in 2–4 mL of a chloroform/methanol mixture (3:7, v/v) within a round-bottom rotary flask [36].
- Step 2: Thin Film Formation. Evaporate the organic solvent using a rotary evaporator under reduced pressure (200–300 mbar) at a temperature of 35–45 °C in a water bath. This deposits a thin lipid film on the inner wall of the flask. For complete solvent removal, further dry the film under high vacuum (5–10 mbar) until a constant weight is achieved [36].
- Step 3: Hydration. Pre-heat the lipid film and an aqueous buffer solution above the transition temperature (Tm) of the lipids. Add the buffer to the flask to achieve a final lipid concentration of 0.5–10 mg/mL. Hydrate the film for 45 minutes with occasional vigorous shaking and brief sonication in an ultrasonic bath (≈30 s/sonication) to detach the film and form MLVs [36].
- Step 4: Post-Processing (Optional). The resulting heterogeneous MLVs can be processed into small or large unilamellar vesicles (SUVs or LUVs) using downsizing techniques such as sonication (using a probe sonicator on ice) or extrusion (passing the MLVs multiple times through a polycarbonate membrane with a defined pore size) [37].
Ethanol Injection
This method is rapid and simple, favoring the formation of unilamellar vesicles with minimal solvent residue concerns [37].
- Step 1: Lipid Dissolution. Dissolve the phospholipids in ethanol [37].
- Step 2: Rapid Injection. Pre-heat an aqueous medium containing the material to be encapsulated. Rapidly inject the ethanolic lipid solution through a needle into the stirred aqueous media [37].
- Step 3: Vesicle Formation and Solvent Removal. The spontaneous self-assembly of lipids into liposomes occurs instantly due to the change in polarity as the ethanol mixes with the aqueous phase. Stir the mixture at an elevated temperature (55–65 °C) to facilitate the evaporation of residual ethanol [37]. The final liposome size (SUVs or LUVs) is influenced by the rate of injection and the volume of ethanol, with volumes not exceeding 7.5% of the total formulation volume favoring homogeneous SUVs [37].
Reverse-Phase Evaporation
This technique is particularly noted for achieving high encapsulation efficiencies for hydrophilic molecules [37].
- Step 1: Emulsion Formation. Dissolve lipids in an organic solvent such as chloroform/methanol (2:1 v/v). Add an aqueous buffer containing the drug to be encapsulated to the lipid solution and sonicate the mixture to form a stable water-in-oil emulsion [37].
- Step 2: Solvent Evaporation. Remove the organic solvent by rotary evaporation under reduced pressure. This process transforms the emulsion first into a viscous gel [37].
- Step 3: Gel Collapse and Liposome Formation. Continued evaporation leads to the collapse of the gel structure, resulting in the formation of a liposomal suspension. The large aqueous core of the original emulsion droplets contributes to the high encapsulation efficiency of this method [37].
The following workflow diagram summarizes the steps of these preparation methods and their resulting liposome types.
Comparative Performance Analysis of Preparation Methods
The choice of preparation method significantly impacts critical quality attributes of the final liposomal formulation. The table below provides a comparative analysis of the three methods based on key performance metrics.
Table 1: Comparative Analysis of Liposome Preparation Methods
| Performance Metric | Thin-Film Hydration | Ethanol Injection | Reverse-Phase Evaporation |
|---|---|---|---|
| Principle | Drying & hydration [37] | Polarity change [37] | Emulsion evaporation [37] |
| Typical Liposome Type | Multilamellar Vesicles (MLVs) [36] [37] | Small/Large Unilamellar Vesicles (SUVs/LUVs) [37] | Large Unilamellar Vesicles (LUVs) [37] |
| Encapsulation Efficiency | Low for hydrophilic drugs [37] | Low for lipophilic drugs [37] | High for hydrophilic molecules [37] |
| Size Control | Requires post-processing (e.g., extrusion) [37] | Moderate (depends on injection rate) [37] | Good [37] |
| Scalability | Difficult to scale up [37] | Good for lab-scale, simple [37] | Challenging, time-consuming [37] |
| Residual Solvent | Chloroform/Methanol (difficult to remove) [37] | Ethanol (less toxic, easier to remove) [37] | Chloroform/Methanol (significant amounts) [37] |
| Key Advantage | Simple, highly reproducible [36] [37] | Rapid, simple process [37] | High aqueous encapsulation [37] |
| Key Disadvantage | Low encapsulation efficiency, heterogeneous initial size [37] | Potential for heterogeneous liposomes, solvent dilution [37] | Uses large amounts of organic solvent [37] |
Beyond these conventional methods, novel techniques like microfluidics are emerging. Microfluidic devices allow for the continuous and controlled production of liposomes with a narrow size distribution by mixing lipid solutions in alcohol with an aqueous buffer in a micromixer chip [37]. While this method offers excellent size control and is suitable for a continuous flow process, challenges remain regarding solvent removal and the cost of cartridge renewal [37].
The Scientist's Toolkit: Essential Research Reagents
Successful liposome formulation relies on a specific set of reagents and instruments. The table below details the essential components of a liposome research toolkit.
Table 2: Key Research Reagents and Materials for Liposome Preparation
| Reagent/Material | Function in Liposome Preparation | Examples & Notes |
|---|---|---|
| Phospholipids | Structural backbone of the lipid bilayer. | DPPC: Saturated lipid with high Tm for rigid bilayers [36].POPC: Unsaturated lipid for more fluid bilayers [36].HSPC/DSPC: Common in commercial products for stability [34]. |
| Cholesterol | Bilayer excipient that stabilizes membrane, reduces permeability, and improves in vivo stability [33]. | Typically used at 20-50 mol% relative to phospholipids [33]. |
| Polyethylene Glycol (PEG)-Lipid | Creates "stealth" liposomes; prolongs circulation by reducing RES uptake [33] [8]. | e.g., MPEG-DSPE. Used in Doxil/Caelyx [34]. |
| Organic Solvents | Dissolve lipids for initial processing. | Chloroform/Methanol: Used in thin-film hydration [36].Ethanol: Used in ethanol injection (Class 3, less toxic) [37]. |
| Hydration Buffer | Aqueous medium for hydrating the lipid film, can contain hydrophilic drugs. | Phosphate-buffered saline (PBS), HEPES, or other buffers at physiological pH and osmolarity [36]. |
| Critical Equipment | Enables formation and sizing of liposomes. | Rotary Evaporator: For solvent removal and thin-film formation [36] [37].Sonicator (Bath/Probe): For downsizing MLVs to SUVs [37].Extruder: For producing homogeneous LUVs/SUVs via membrane extrusion [36] [37]. |
The selection of an optimal liposome preparation method is a critical decision that balances experimental needs with practical constraints. Thin-film hydration remains the gold standard for its simplicity and reproducibility, ideal for initial formulation development despite its need for post-processing sizing. Ethanol injection offers a rapid, straightforward path to unilamellar vesicles with lower solvent toxicity. Reverse-phase evaporation is the method of choice when high encapsulation efficiency for hydrophilic drugs is the primary objective, though it comes with the burden of higher solvent use.
When framing this choice within the broader context of nanocarrier research, the distinct advantages of liposomes—their proven clinical track record and high biocompatibility—are counterbalanced by the potentially superior stability and highly tunable drug release profiles offered by polymeric nanoparticles [38] [1]. Ultimately, the research question, the physicochemical properties of the active compound, and the intended therapeutic application will guide not only the choice of preparation method but also the fundamental selection between liposomal and polymeric nanocarrier platforms. Future advancements will likely focus on hybrid techniques and scalable technologies like microfluidics to overcome current limitations in encapsulation efficiency, solvent residue, and industrial manufacturability [33] [37].
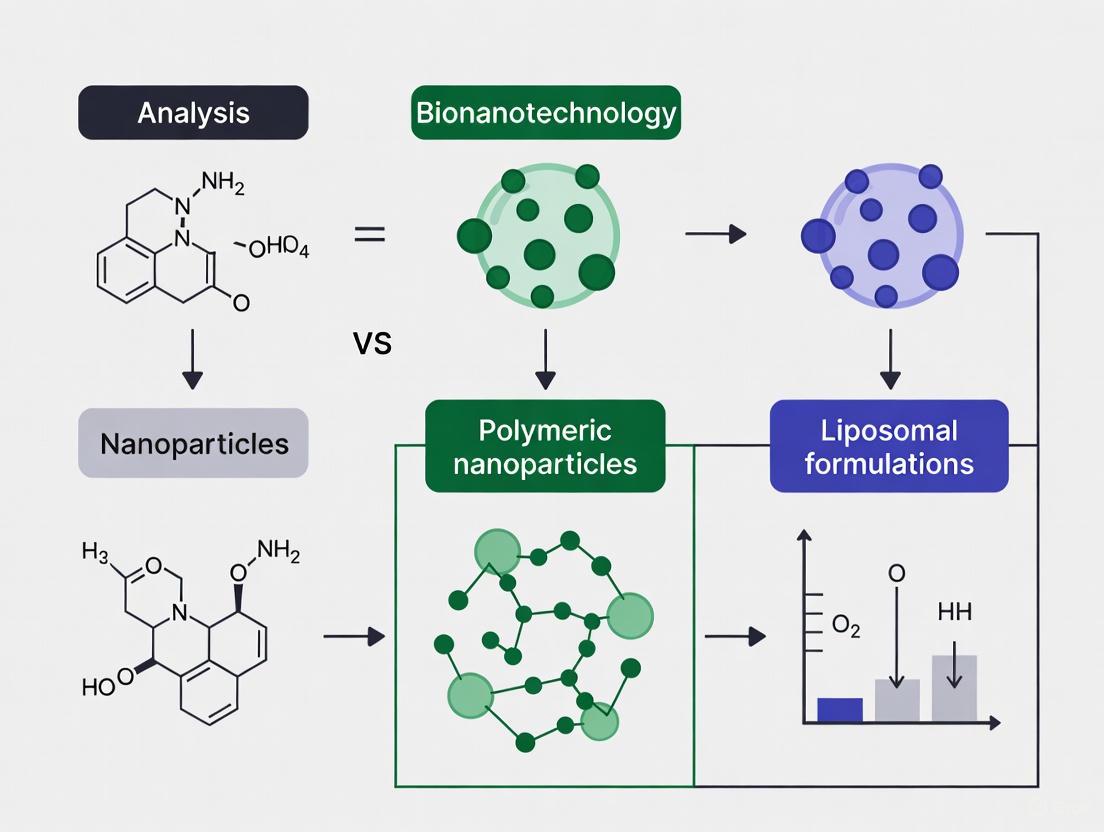
In modern therapeutics, nanocarriers have revolutionized drug delivery by improving the pharmacokinetics and safety profiles of active pharmaceutical ingredients. Among the most prominent of these carriers are liposomal formulations and polymeric nanoparticles, which offer distinct advantages and challenges [8] [39]. Liposomes are spherical phospholipid vesicles with structural similarities to biological membranes, enabling encapsulation of both hydrophilic and hydrophobic drugs [23] [40]. Polymeric nanoparticles, conversely, are typically composed of biodegradable polymers that can provide robust control over drug release kinetics [39]. This review provides a comprehensive comparative analysis of these two platforms, focusing on three critical engineering strategies for enhanced performance: PEGylation for stealth properties, stimuli-responsive mechanisms for controlled release, and active targeting ligands for precision delivery. The objective assessment of experimental data and performance metrics presented herein offers valuable insights for researchers selecting appropriate nanocarrier systems for specific therapeutic applications.
Comparative Analysis of Liposomal and Polymeric Nanoparticle Platforms
Table 1: Fundamental characteristics of liposomal versus polymeric nanoparticle platforms
| Characteristic | Liposomal Formulations | Polymeric Nanoparticles |
|---|---|---|
| Structural Composition | Phospholipid bilayers (natural/synthetic) with cholesterol [40] | Biodegradable polymers (e.g., PLGA, PEG-PLGA) [39] |
| Architecture | Unilamellar or multilamellar vesicles enclosing aqueous core [23] | Solid matrix or core-shell structures (micelles) [39] |
| Drug Encapsulation | Hydrophilic drugs in aqueous core; hydrophobic drugs in lipid bilayer [23] | Primarily hydrophobic drugs in polymer matrix; conjugation approaches for hydrophilic drugs [39] |
| Biocompatibility | High (biomimetic cellular membrane structure) [40] | Variable (depends on polymer composition) [39] |
| Manufacturing Complexity | Moderate (challenges with batch-to-batch uniformity) [23] | Moderate to high (requires polymer synthesis expertise) [39] |
| Regulatory Approval Status | Multiple FDA-approved products [40] | Limited approved products, though platforms are well-established [39] |
Table 2: Drug loading capacity and encapsulation efficiency comparison
| Parameter | Liposomal Formulations | Polymeric Nanoparticles |
|---|---|---|
| Typical Loading Methods | Passive loading, active/remote loading using pH gradients [23] | Nanoprecipitation, emulsification-solvent evaporation [41] |
| Loading Capacity Challenges | Limited for conventional liposomes; improved with advanced strategies [23] | Generally high for hydrophobic drugs; lower for hydrophilic compounds [39] |
| Encapsulation Efficiency | Variable (5-90% depending on method and drug properties) [23] | Typically moderate to high (50-90% for compatible drugs) [39] |
| Strategies for Enhancement | Transmembrane ion gradients, lipid composition optimization, supercritical fluid technologies [23] | Polymer-drug conjugation, surface modification, hybrid systems [39] |
The fundamental structural differences between liposomal and polymeric nanoparticle systems directly influence their drug encapsulation capabilities and biological performance. Liposomes offer a unique advantage with their ability to encapsulate both water-soluble compounds within their aqueous core and lipid-soluble drugs within their bilayer membrane [23]. This amphiphilic loading capacity makes them particularly versatile for combination therapies where drugs with different solubility profiles must be delivered simultaneously. Polymeric nanoparticles, typically formed from materials such as PLGA, poly(alkyl cyanoacrylates), or chitosan, generally excel at encapsulating hydrophobic compounds within their solid polymer matrix, though surface modification and conjugation strategies can expand their loading capabilities [39].
PEGylation: Enhancing Circulation Time and Stability
Polyethylene glycol (PEG) conjugation represents a cornerstone strategy for improving the pharmacokinetic profiles of both liposomal and polymeric nanocarriers. This approach involves attaching PEG chains to the nanoparticle surface to create a "stealth" effect that reduces recognition and clearance by the mononuclear phagocyte system (MPS) [8] [41].
PEGylation Techniques and Experimental Considerations
Table 3: PEGylation approaches for nanocarrier systems
| PEGylation Method | Mechanism | Applicability | Experimental Considerations |
|---|---|---|---|
| Covalent Grafting | Stable chemical bond formation between activated PEG and nanoparticle surface [41] | Both liposomal and polymeric systems | Requires functional groups on nanoparticle surface; must characterize binding efficiency |
| Physical Adsorption | Electrostatic or hydrophobic interactions between PEG-containing molecules and nanoparticle [41] | More common for polymeric systems | Less stable than covalent approaches; sensitive to environmental conditions |
| PEG-Lipid Incorporation | Anchoring PEG-lipid conjugates during liposome formation [8] | Primarily liposomal systems | PEG-lipid must be included during formulation; can affect membrane properties |
| Self-Assembly | PEG-coupled compounds partition during nanoparticle formation [41] | Both systems, particularly polymeric | Method depends on nanoprecipitation or emulsification techniques |
The experimental protocol for PEGylation typically involves synthesizing or procuring activated PEG derivatives with appropriate functional groups (e.g., PEG-NHS for amine coupling, PEG-MAL for thiol coupling). For liposomal systems, PEG-lipid conjugates (such as DSPE-PEG) are incorporated during the thin-film hydration or microfluidic mixing processes at concentrations typically ranging from 1-10 mol% relative to total lipids [8]. For polymeric nanoparticles, PEG can be incorporated as a block copolymer (e.g., PLGA-PEG) during the nanoprecipitation or emulsion process, or through post-synthesis "grafting-to" approaches where activated PEG is reacted with functional groups on pre-formed nanoparticles [41]. Critical quality control measures include determining PEG density on the nanoparticle surface through colorimetric assays or NMR spectroscopy, measuring zeta potential changes, and evaluating the reduction in protein binding through serum incubation studies.
Comparative Performance of PEGylated Systems
Table 4: Quantitative comparison of PEGylation effects on nanocarrier performance
| Performance Metric | PEGylated Liposomes | PEGylated Polymeric Nanoparticles | Experimental Evidence |
|---|---|---|---|
| Circulation Half-life | 20-80 hours (vs. 2-4 hours for conventional) [8] | 12-48 hours (variable based on polymer) [39] | Pharmacokinetic studies in rodent models |
| Protein Adsorption Reduction | Up to 80% reduction in opsonin binding [8] | 60-90% reduction (depends on PEG density) [41] | Serum protein binding assays; 2D electrophoresis |
| Tumor Accumulation (EPR Effect) | 5-15% injected dose/g tissue [8] | 3-10% injected dose/g tissue [39] | Radiolabeling studies in xenograft models |
| Immunogenicity Concerns | ABC phenomenon with anti-PEG IgM [42] [43] | Similar ABC phenomenon observed [42] | Repeated administration studies measuring clearance rates |
PEGylation Benefits and Limitations Flowchart: Visual representation of the opposing effects of PEGylation on nanocarrier performance, highlighting the balance between extended circulation and potential drawbacks.
While PEGylation significantly enhances circulation time for both nanoparticle platforms, important limitations must be considered. The "accelerated blood clearance" (ABC) phenomenon, where subsequent doses of PEGylated nanoparticles are cleared more rapidly due to anti-PEG antibody production, presents a challenge for chronic therapy regimes with both systems [42] [43]. Additionally, the steric hindrance created by PEG chains can potentially reduce cellular uptake and intracellular delivery, particularly for gene therapies where endosomal escape is crucial [43]. The optimal PEG configuration—including molecular weight (typically 2-5 kDa for liposomes, 2-20 kDa for polymeric systems), chain density (5-10% molar ratio for liposomes), and architecture (linear versus branched)—must be empirically determined for each specific nanocarrier and therapeutic application [8] [41].
Stimuli-Responsive Systems: Controlled Drug Release at Target Sites
Stimuli-responsive nanocarriers represent an advanced generation of drug delivery systems designed to release their payload in response to specific physiological or external triggers. These systems maximize therapeutic efficacy while minimizing off-target effects through spatiotemporal control of drug release [44].
Endogenous Stimuli-Responsive Mechanisms
Table 5: Endogenous stimuli-responsive systems for targeted drug release
| Stimulus Type | Mechanism | Liposomal Approaches | Polymeric Approaches |
|---|---|---|---|
| pH-Responsive | Drug release in acidic environments (tumors, endosomes, inflamed tissues) [23] | pH-sensitive lipids (e.g., DOPE); acid-labile linkers [23] | Polymers with ionizable groups; acid-cleavable linkers [44] |
| Redox-Responsive | Cleavage in high glutathione environments (cytoplasm, tumors) [44] | Thiol-responsive phospholipids; disulfide bridges [40] | Disulfide-crosslinked polymers; thioketal-based systems [44] |
| Enzyme-Responsive | Substrate cleavage by disease-associated enzymes [44] | Enzyme-sensitive lipid conjugates; peptide-based linkers [8] | Enzyme-degradable polymer backbones; substrate-conjugated systems [44] |
| Hypoxia-Responsive | Activation in low oxygen environments (tumors) [44] | Nitroimidazole-conjugated lipids; azobenzene linkers [44] | Quinone-based polymers; azobenzene-containing systems [44] |
The experimental methodology for evaluating stimuli-responsive systems typically involves in vitro release studies under conditions mimicking the pathological environment. For pH-responsive systems, drug release is quantified using dialysis methods at progressively decreasing pH values (e.g., 7.4, 6.5, 5.5) with samples analyzed via HPLC or spectrophotometry at predetermined time points [23]. Redox-responsive release is assessed by adding reducing agents such as glutathione or dithiothreitol to the release medium, with concentrations typically ranging from 10 μM (extracellular) to 10 mM (intracellular) [44]. Enzyme-responsive systems require incubation with specific enzymes (e.g., matrix metalloproteinases, esterases) at physiological concentrations, with release kinetics compared to control conditions without enzymes or with enzyme inhibitors [8].
Comparative Performance of Stimuli-Responsive Systems
Table 6: Quantitative comparison of stimuli-responsive nanocarrier performance
| Performance Metric | Stimuli-Responsive Liposomes | Stimuli-Responsive Polymeric Nanoparticles | Experimental Models |
|---|---|---|---|
| Triggered Release Efficiency | 3-5 fold increase vs. non-responsive [23] | 4-8 fold increase vs. non-responsive [44] | In vitro release studies under simulated conditions |
| Tumor Growth Inhibition | 60-80% reduction vs. 30-50% for conventional [23] | 70-90% reduction vs. 40-60% for conventional [44] | Xenograft mouse models (e.g., MDA-MB-231, HT-29) |
| Therapeutic Index Improvement | 2-3 fold increase [23] | 3-5 fold increase [44] | Ratio of efficacy to toxicity in preclinical models |
| Target/Non-Target Ratio | 3-8 fold higher accumulation in target tissue [23] | 5-10 fold higher accumulation in target tissue [44] | Biodistribution studies with radiolabeled carriers |
Stimuli-Responsive Trigger Mechanisms: Overview of the different trigger types that can initiate controlled drug release from responsive nanocarriers.
Beyond endogenous triggers, exogenous stimuli-responsive systems activated by external energy sources offer precise spatiotemporal control. Thermo-responsive liposomes incorporating temperature-sensitive lipids (such as DPPC) release their payload upon mild hyperthermia (39-42°C) at the target site [23]. Similarly, thermo-responsive polymeric systems using polymers like poly(N-isopropylacrylamide) exhibit phase transitions at specific temperatures to modulate drug release [39]. Ultrasound-responsive systems, particularly valuable for deep tissue applications, utilize microbubble liposomes or echogenic polymers that undergo cavitation or mechanical disruption upon ultrasound exposure, typically at frequencies of 1-3 MHz and intensities of 1-3 W/cm² [40]. Light-responsive systems, while offering exceptional precision, are generally limited to superficial applications due to tissue penetration constraints [44].
Active Targeting Ligands: Precision Delivery to Specific Cells
Active targeting represents the third pillar of nanocarrier engineering, involving the conjugation of specific ligands to the nanoparticle surface to facilitate receptor-mediated binding and internalization by target cells [8].
Targeting Ligand Conjugation Strategies
The experimental protocol for ligand conjugation typically involves incorporating functionalized lipids (e.g., DSPE-PEG-MAL, DSPE-PEG-NHS) into liposomal formulations or creating functional groups on polymeric nanoparticles for subsequent ligand attachment. Common conjugation approaches include thiol-maleimide chemistry, carbodiimide-mediated amide bond formation, and click chemistry reactions [8]. For example, the methodology for conjugating monoclonal antibodies or their fragments to PEGylated nanocarriers involves first introducing maleimide groups at the distal end of PEG chains, followed by reaction with thiolated targeting ligands at controlled pH (6.5-7.5) to minimize disulfide formation. The conjugation efficiency is typically quantified using colorimetric assays, HPLC, or mass spectrometry, with the optimal ligand density determined through binding studies with target cells [8].
Comparative Performance of Actively Targeted Systems
Table 7: Experimentally demonstrated targeting ligands and their efficacy
| Ligand Type | Target Receptor | Nanocarrier Platform | Targeting Efficiency Improvement |
|---|---|---|---|
| Monoclonal Antibodies | HER2, EGFR, CD20 [8] | Both liposomal and polymeric | 5-15 fold increase in cellular uptake [8] |
| Peptides (RGD, TAT) | Integrins, cell penetration [8] | Primarily liposomal | 3-8 fold increase in tumor accumulation [8] |
| Aptamers | Various cancer markers [41] | Both platforms | 4-10 fold binding affinity increase [41] |
| Small Molecules (Folate) | Folate receptor [8] | Both platforms | 5-12 fold increase in internalization [8] |
Table 8: In vivo performance comparison of targeted versus non-targeted systems
| Performance Metric | Actively Targeted Liposomes | Actively Targeted Polymeric Nanoparticles | Experimental Evidence |
|---|---|---|---|
| Cellular Uptake | 3-8 fold increase vs. non-targeted [8] | 4-10 fold increase vs. non-targeted [39] | Flow cytometry; confocal microscopy |
| Tumor Growth Inhibition | 70-90% vs. 40-60% for non-targeted [8] | 75-95% vs. 45-65% for non-targeted [39] | Xenograft models measuring tumor volume |
| Survival Benefit | 40-60% increase in median survival [8] | 50-70% increase in median survival [39] | Animal survival studies |
| Target/Non-Target Ratio | 5-15 fold selective accumulation [8] | 8-20 fold selective accumulation [39] | Biodistribution studies |
The critical consideration in active targeting is balancing the benefits of enhanced cellular uptake against potential complications. While targeted systems demonstrate significantly improved cellular internalization, they may also experience accelerated clearance from circulation due to increased interactions with the reticuloendothelial system [8]. Additionally, the heterogeneity of target receptor expression within diseased tissues, particularly tumors, can limit the universal efficacy of targeted approaches. The optimal ligand density must be empirically determined, as excessive conjugation can compromise stealth properties and circulation time, while insufficient density fails to provide meaningful targeting benefits [8].
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Table 9: Essential research reagents for advanced nanocarrier development
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| PEGylation Reagents | DSPE-PEG (1k-5k Da), PLGA-PEG, methoxy-PEG-NHS [41] | Stealth properties, circulation extension | Molecular weight affects clearance; density must be optimized |
| pH-Sensitive Materials | DOPE, CHEMS, poly(histidine) derivatives [23] | Endosomal escape; tumor microenvironment targeting | pKa determines activation pH; must match target environment |
| Targeting Ligands | Folate, RGD peptides, trastuzumab fragments, transferrin [8] | Specific cell recognition and internalization | Binding affinity must be balanced with immunogenicity |
| Characterization Tools | Dynamic light scattering, HPLC, cryo-TEM [23] | Size, stability, and encapsulation efficiency | Multiple complementary techniques required for full characterization |
The development of advanced nanocarrier systems requires specialized reagents and methodologies. For PEGylation, DSPE-PEG derivatives remain the gold standard for liposomal systems, with molecular weights typically ranging from 1,000 to 5,000 Da [41]. For polymeric nanoparticles, PLGA-PEG diblock or triblock copolymers enable the formation of sterically stabilized particles with controlled release properties [39]. In stimuli-responsive formulations, pH-sensitive lipids such as DOPE (dioleoylphosphatidylethanolamine) are commonly employed in liposomal systems, while poly(histidine) and acetal-based polymers serve similar functions in polymeric platforms [23] [44]. Targeting ligand conjugation requires heterobifunctional crosslinkers such as DSPE-PEG-MAL or NHS-PEG-MAL that first anchor to the nanoparticle surface then provide functional groups for ligand attachment [8].
Critical experimental protocols for evaluating these advanced systems include:
- Microfluidic fabrication: For both liposomal and polymeric systems, microfluidic approaches enable precise control over nanoparticle size and polydispersity [45]. Typical setups involve staggered herringbone mixers or hydrodynamic flow focusing geometries with aqueous and organic phases controlled by syringe pumps at flow rates ranging from 1-10 mL/min.
- Stability assessment: Accelerated stability studies conducted at 4°C, 25°C, and 37°C in physiologically relevant media (PBS, serum) over 1-3 months, with monitoring of size (DLS), zeta potential, drug retention (HPLC), and visual appearance.
- In vitro efficacy testing: Cell culture models including 2D monolayers and 3D spheroids, with cytotoxicity assessed via MTT or PrestoBlue assays, cellular uptake quantified by flow cytometry or confocal microscopy, and internalization mechanisms probed using endocytic inhibitors.
- In vivo evaluation: Biodistribution studies using radiolabeled (e.g., 111In, 99mTc) or fluorescently labeled (DiR, Cy7) nanoparticles in appropriate disease models, with therapeutic efficacy assessed through tumor volume reduction, survival extension, or biomarker modulation.
The comprehensive comparison presented herein demonstrates that both liposomal and polymeric nanoparticle platforms offer distinct advantages for drug delivery applications. Liposomal systems benefit from their biomimetic structure, clinical validation, and versatility in encapsulating diverse therapeutic agents [40]. Polymeric nanoparticles provide superior control over drug release kinetics and potential for higher drug loading of compatible compounds [39]. The engineering strategies of PEGylation, stimuli-responsiveness, and active targeting significantly enhance the performance of both platforms, though optimal implementation requires careful consideration of the trade-offs involved. PEGylation extends circulation but may induce immunogenicity and reduce cellular uptake [42] [43]. Stimuli-responsive systems enable precise drug release but add formulation complexity [44]. Active targeting enhances specificity but must be balanced against potential accelerated clearance [8]. The selection between liposomal and polymeric platforms should be guided by the specific therapeutic application, drug properties, manufacturing considerations, and regulatory pathway. Future directions will likely focus on hybrid systems that combine advantageous elements of both platforms, along with increasingly sophisticated bioresponsive mechanisms that adapt to complex disease microenvironments.
In the evolving landscape of nanomedicine, polymeric nanoparticles (PNPs) and liposomal formulations have emerged as leading drug delivery systems. While both leverage nanotechnology to improve therapeutic outcomes, their distinct structural compositions, material properties, and interaction mechanisms with biological systems define their specific application niches and performance. This guide provides a comparative analysis of PNPs and liposomes across three key therapeutic areas: oncology, neurological disorders, and dermatology. It is structured to offer researchers, scientists, and drug development professionals an objective, data-driven resource, complete with experimental data, methodologies, and essential research tools to inform platform selection and development.
Comparative Analysis in Oncology: Exploiting the EPR Effect
The Enhanced Permeability and Retention (EPR) effect is a cornerstone of nanomedicine in oncology, allowing nanocarriers to accumulate preferentially in tumor tissues. Both PNPs and liposomes utilize this phenomenon, but their structural differences lead to variations in drug loading, release kinetics, and tumor microenvironment (TME) interaction.
Table 1: Comparison of PNPs and Liposomes in Oncology Applications
| Feature | Polymeric Nanoparticles (PNPs) | Liposomal Formulations |
|---|---|---|
| Material Composition | Biodegradable polymers (e.g., PLGA, chitosan, gelatin, polycaprolactone) [46]. | Phospholipid bilayers (e.g., phosphatidylcholine), cholesterol [47] [8]. |
| Structure | Solid, monolithic matrix [46]. | Aqueous core enclosed by one or more lipid bilayers [8]. |
| Drug Loading | High encapsulation efficiency for both hydrophilic and hydrophobic drugs; controlled via polymer composition and degradation [46]. | Hydrophilic drugs in aqueous core; hydrophobic drugs in lipid bilayers [47] [8]. |
| Release Kinetics | Sustained and controlled release, tuned by polymer degradation rate [46]. | Rapid initial burst release, followed by sustained release; can be tuned with lipid composition and PEGylation [8]. |
| TME Interaction | Engineered for stimuli-responsive release (e.g., pH, enzymes). Can co-deliver immunomodulators to remodel TME [46]. | Passive targeting via EPR effect. Stealth (PEGylated) versions evade immune system for longer circulation [46] [8]. |
| Key Advantages | High stability, tunable degradation, and potential for sophisticated TME modulation [46]. | Excellent biocompatibility, proven clinical translation (e.g., Doxil), and high drug payload [8]. |
| Reported Tumor Inhibition (Preclinical) | ~60-80% reduction in tumor volume in ligand-targeted and stimuli-responsive PNP models in lung cancer [46]. | ~50-70% tumor growth inhibition with PEGylated liposomal doxorubicin, compared to ~20-30% for conventional chemotherapy [46]. |
Experimental Spotlight: PNPs for Modulating the Lung Cancer Microenvironment
- Objective: To evaluate the efficacy of multifunctional PNPs in delivering chemotherapeutic agents and modulating the TME to overcome drug resistance in lung cancer [46].
- Methodology:
- NP Fabrication: PNPs were formulated using poly(lactic-co-glycolic acid) (PLGA) via a nano-precipitation method. The NPs were co-loaded with a chemotherapeutic agent (e.g., docetaxel) and an immunomodulator (e.g., a STAT3 inhibitor) [46].
- Surface Functionalization: The PNPs were conjugated with ligands (e.g., folic acid or peptides) targeting receptors overexpressed on lung cancer cells to enhance active targeting [46].
- In Vitro Evaluation: Cellular uptake, cytotoxicity, and apoptosis assays were performed on human non-small cell lung cancer (NSCLC) cell lines. The ability of PNPs to disrupt pro-survival signaling pathways (e.g., STAT3) was analyzed via western blotting [46].
- In Vivo Evaluation: An orthotopic lung cancer mouse model was used. Animals were treated intravenously with saline, free drug, non-targeted PNPs, and ligand-targeted PNPs. Tumor volume was monitored via bioluminescence imaging. Analysis of tumor tissues post-treatment assessed drug concentration, hypoxia markers (HIF-1α), and immune cell infiltration [46].
- Key Outcome: Ligand-conjugated, multi-agent PNPs demonstrated superior tumor accumulation and penetration, leading to a significant reduction in tumor volume (~70-80%) compared to free drug or non-targeted NPs. The combination therapy effectively suppressed immunosuppressive signals and reduced hypoxia, sensitizing the tumor to chemotherapy [46].
Diagram 1: Mechanism of targeted polymeric nanoparticles (PNPs) in cancer therapy, showing the journey from injection to tumor cell death and microenvironment (TME) modulation.
Comparative Analysis in Neurological Disorders: Crossing the Blood-Brain Barrier
The blood-brain barrier (BBB) is a major obstacle in treating neurological diseases. Both PNPs and liposomes can be engineered for brain targeting, primarily utilizing receptor-mediated transcytosis (RMT) and adsorptive-mediated transcytosis (AMT).
Table 2: Comparison of PNPs and Liposomes in Crossing the BBB for Neurological Applications
| Feature | Polymeric Nanoparticles (PNPs) | Liposomal Formulations |
|---|---|---|
| Material Composition | Synthetic (e.g., PLGA) or natural (e.g., chitosan) polymers [46] [48]. | Phospholipids, cholesterol, PEG-lipids [49] [8]. |
| Common Targeting Ligands | Peptides (e.g., TAT), transferrin (Tf), antibodies [50] [48]. | Transferrin (Tf), cell-penetrating peptides (e.g., Penetratin), glutathione [49] [8]. |
| Primary Transcytosis Mechanism | Receptor-mediated (RMT) and Adsorptive-mediated (AMT) [50] [48]. | Receptor-mediated (RMT) [49] [8]. |
| Payload Flexibility | Genes, small molecules, proteins; controlled release profile [46]. | Genes, small molecules; good for nucleic acids due to fusion with cell membranes [49] [8]. |
| Key Advantages | Rigid structure allows precise surface engineering and high stability. Dendrimers offer exceptional functionalization control [50]. | Innate biocompatibility and fluid bilayer facilitates membrane fusion and endosomal escape [49] [47]. |
| Reported Efficacy (Preclinical) | Dendrimer-based NPs show significant improvement in behavioral outcomes and reduction in amyloid plaques in Alzheimer's models [50]. | Tf/Penetratin-dual functionalized liposomes increased brain ACE2 expression by >5-fold, attenuating neurogenic hypertension in rats [49]. |
Experimental Spotlight: Dual-Functionalized Liposomes for Neurogenic Hypertension
- Objective: To develop a liposome system for delivering the human angiotensin-converting enzyme 2 (ACE2) gene across the BBB to treat neurogenic hypertension [49].
- Methodology:
- Liposome Preparation: PEGylated liposomes were prepared using a thin-film hydration method, composed of DOPE, DOTAP, and cholesterol. Plasmid DNA encoding ACE2 (pACE2) was condensed with chitosan and encapsulated [49].
- Surface Functionalization: Liposomes were dual-modified with Holo-Transferrin (Tf) and the cell-penetrating peptide Penetratin (Pen). Tf was conjugated via DSPE-PEG2000-NHS chemistry to target Tf receptors on the BBB, while Pen was incorporated to enhance cellular uptake and endosomal escape [49].
- In Vitro BBB Model: Transport efficiency was quantified using a co-culture model of brain endothelial cells (bEnd.3), astrocytes, and pericytes. The integrity of the barrier was monitored by measuring transendothelial electrical resistance (TEER) [49].
- In Vivo Evaluation: Sprague-Dawley rats with angiotensin II-induced neurogenic hypertension were administered Tf-Pen-Lip-pACE2 intravenously. Gene expression in the hypothalamic paraventricular nucleus (PVN) was confirmed by immunohistochemistry. Blood pressure and heart rate were monitored chronically. Safety was assessed via plasma biomarkers of liver and kidney function [49].
- Key Outcome: The Tf-Pen-Lip system successfully delivered the pACE2 gene across the BBB, significantly increasing ACE2 expression in the PVN. This treatment dramatically attenuated the hypertension and sympathetic overactivity induced by Ang II, without evident systemic toxicity [49].
Diagram 2: Two primary mechanisms for nanoparticle transport across the blood-brain barrier (BBB): receptor-mediated transcytosis (RMT) and adsorptive-mediated transcytosis (AMT).
Comparative Analysis in Dermatological Treatments
The skin barrier presents a unique delivery challenge. Lipid-based systems like liposomes have a natural affinity for the stratum corneum, while PNPs offer superior stability and controlled release for deeper skin conditions.
Table 3: Comparison of PNPs and Liposomes in Dermatological Applications
| Feature | Polymeric Nanoparticles (PNPs) | Liposomal Formulations |
|---|---|---|
| Material Composition | PLGA, chitosan, gelatin [46] [51]. | Phospholipids, often in hybrid Lipid-Polymer Nanoparticles (LPN) [52]. |
| Skin Penetration | Can be engineered for follicular delivery or enhanced penetration into deeper skin layers [51]. | Primarily localizes in the stratum corneum; excellent for superficial delivery and moisturization [53]. |
| Drug Release | Sustained release over days to weeks, ideal for chronic conditions [51]. | Faster release, suitable for providing an immediate drug reservoir on the skin [53]. |
| Formulation Versatility | Can be incorporated into gels, creams, and patches. Tiny size avoids rapid mucus clearance in pulmonary delivery for skin cancer metastasis [46]. | Highly versatile; used in creams, lotions, and sprays. LPNs combine stability of polymers with biocompatibility of lipids [52]. |
| Key Advantages | High physical and chemical stability, protection of encapsulated drugs, and tunable release kinetics [51]. | Excellent biocompatibility and biomimetic properties, enhancing skin hydration and integrity [53]. |
| Reported Efficacy | PNPs showed enhanced efficacy and reduced side effects in treating psoriasis and atopic dermatitis in preclinical models [51]. | Liposomal formulations of corticosteroids and vitamins demonstrated improved symptom relief and skin hydration in clinical settings for eczema and psoriasis [53]. |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Reagents for Nanoparticle Formulation and Testing
| Reagent / Material | Function | Example Application |
|---|---|---|
| PLGA | A biodegradable copolymer forming the core matrix of PNPs; degradation rate tuned by lactic to glycolic acid ratio [46]. | Core polymer for sustained-release PNPs in oncology and dermatology [46] [51]. |
| DSPE-PEG2000-NHS | PEGylated phospholipid used for "stealth" functionalization; NHS group allows covalent conjugation to targeting ligands [49] [8]. | Conjugating transferrin or peptides to liposomes/PNPs for active targeting and prolonged circulation [49]. |
| Holo-Transferrin | A targeting ligand that binds to the transferrin receptor (TfR), highly expressed on BBB endothelial cells [49]. | Facilitating receptor-mediated transcytosis across the BBB in neurological disorder research [49]. |
| Penetratin | A cell-penetrating peptide (CPP) that enhances cellular uptake and endosomal escape of nanocarriers [49]. | Improving intracellular delivery of genes and drugs in brain-targeted liposomes [49]. |
| Cholesterol | A stabilizing agent incorporated into lipid bilayers to reduce membrane fluidity and prevent drug leakage [47] [8]. | A key component of liposomal formulations to enhance in vitro and in vivo stability [47]. |
| Chitosan | A natural polysaccharide polymer used for NP formation or DNA complexation; offers mucoadhesive properties [49]. | Forming PNPs for dermatology or condensing DNA in liposomal-polyplex hybrid systems for gene delivery [49] [51]. |
Navigating Hurdles: From Scalability and Stability to Regulatory Pathways
In modern drug delivery, nanocarrier systems like liposomes and polymeric nanoparticles (PNPs) have revolutionized therapeutic strategies for complex diseases. A critical challenge in their development, however, lies in ensuring physical and chemical stability during storage and after administration. Instability can manifest as drug leakage, lipid oxidation, or polymer degradation, directly compromising therapeutic efficacy and safety [10]. This guide provides a comparative analysis of liposomal and polymeric nanoparticle formulations, focusing on their inherent stability challenges, supported by experimental data and methodologies relevant to researchers and drug development professionals.
The core instability issues stem from the fundamental differences in the composition and structure of liposomes and polymeric nanoparticles.
Liposomes, with their phospholipid bilayers, are particularly susceptible to chemical degradation such as lipid oxidation and physical instability leading to drug leakage or fusion [54] [10]. Their aqueous core and lipid membrane structure, while excellent for encapsulating diverse drug types, can be disrupted by environmental factors like pH and enzymes.
Polymeric Nanoparticles, typically composed of polymers like PLA (polylactic acid) or PLGA (polylactic-co-glycolic acid), face challenges related to polymer hydrolysis and colloidal aggregation in biological fluids [55] [22]. Their solid matrix, though often providing superior drug loading, can be vulnerable to chemical degradation that alters release profiles and nanoparticle integrity.
Table 1: Core Instability Mechanisms in Liposomes and Polymeric Nanoparticles
| Instability Type | Liposomes | Polymeric Nanoparticles (e.g., PLA/PLGA) |
|---|---|---|
| Primary Chemical Instability | Lipid oxidation [10] | Polymer hydrolysis/degradation [55] |
| Primary Physical Instability | Drug leakage, fusion, aggregation [54] [10] | Aggregation in biological fluids, drug burst release [55] |
| Key Influencing Factors | Lipid composition, storage temperature, surface charge [56] | Polymer crystallinity, molecular weight, presence of stabilizers [55] [57] |
Quantitative Stability and Performance Data
Direct comparative studies and individual formulation analyses reveal distinct performance characteristics. The data below highlights how formulation choices impact key physicochemical properties.
Table 2: Comparative Physicochemical Properties of L-Carnitine Formulations [11]
| Parameter | L-Carnitine Solution | Lipo-Carnitine (Liposome) | Nano-Carnitine (PLGA Nanoparticle) |
|---|---|---|---|
| Particle Size (nm) | N/A | 97.88 ± 2.96 | 250.90 ± 6.15 |
| Polydispersity Index (PDI) | N/A | 0.35 ± 0.01 | 0.22 ± 0.03 |
| Zeta Potential (mV) | N/A | 6.36 ± 0.54 | -32.80 ± 2.26 |
| Encapsulation Efficiency (%) | N/A | 14.26 ± 3.52 | 21.93 ± 4.17 |
| Drug Release (90% Content) | 1 hour | Maintained controlled release for 12 hours | Maintained controlled release for 12 hours |
Table 3: Stabilizer Impact on Polymeric Nanoparticle Properties [57] This study formulated PLGA-loaded paclitaxel nanoparticles with different stabilizers.
| Stabilizer | Concentration | Resulting Particle Size (nm) | Polydispersity Index (PDI) | Zeta Potential (mV) |
|---|---|---|---|---|
| Pluronic F-127 | 1% | 190 ± 12.42 | 0.13 ± 0.02 | -40.4 ± 1.6 |
| Polyvinyl Alcohol (PVA) | 1% | 210 ± 10.50 | 0.15 ± 0.01 | -35.2 ± 1.8 |
| Poloxamer 407 | 1% | 205 ± 11.20 | 0.14 ± 0.02 | -38.1 ± 1.7 |
Experimental Protocols for Stability Assessment
Robust experimental protocols are essential for characterizing and validating the stability of nano-formulations.
Assessing Colloidal Stability in Biological Fluids
This protocol is critical for predicting the in vivo behavior of nanoparticles and their propensity to aggregate [55].
- Method: Dynamic Light Scattering (DLS) and Spectrophotofluorimetric (SPF) analysis.
- Procedure:
- Prepare Nanoparticle Suspensions: Synthesize PNPs (e.g., PLA, PMMA) or liposomes.
- Mix with Biological Fluids: Combine NP suspensions with relevant biological fluids (e.g., gastric juice, intestinal fluid, serum, tissue homogenates) in a 1:1 (v/v) ratio.
- Incubate and Measure: Incubate the mixtures at 37°C. Monitor changes in particle size distribution over time using DLS.
- Ex vivo Validation: Use SPF analysis as a complementary method to qualitatively confirm aggregation, especially for ex vivo samples.
- Application: This test can show, for example, that PLA NPs aggregate in gastric juice and spleen homogenate, while PMMA NPs remain stable, informing the selection of polymer for a given administration route [55].
In Vitro Drug Release Kinetics Study
This standard protocol evaluates the controlled-release profile and potential for premature drug leakage.
- Method: Dialysis or Franz diffusion cell apparatus under sink conditions.
- Procedure:
- Place Formulation: Introduce a known quantity of the nano-formulation (e.g., L-carnitine-loaded liposome or PNP) into a dialysis membrane bag or the donor chamber.
- Immerse in Release Medium: Place the setup in a vessel containing a suitable buffer (e.g., phosphate-buffered saline, pH 7.4) maintained at 37°C under continuous stirring.
- Sample and Analyze: At predetermined time intervals, withdraw samples from the release medium and replace with fresh buffer to maintain sink conditions.
- Quantify Drug Content: Analyze the samples using HPLC or UV-Vis spectroscopy to determine the cumulative drug release over time.
- Application: This method demonstrated that while free L-carnitine releases 90% within an hour, both lipo- and nano-carnitine maintained a controlled release for up to 12 hours [11].
Degradation Pathways and Stabilization Strategies
The degradation mechanisms for liposomes and polymeric nanoparticles are distinct, requiring different stabilization approaches. The following diagrams illustrate these primary pathways and key mitigation strategies.
Liposome Degradation and Stabilization
Liposome instability is driven by chemical and physical pathways. Chemically, phospholipids undergo lipid oxidation and ester bond hydrolysis, compromising membrane integrity [10]. Physically, drugs can leak from the bilayer or aqueous core, and vesicles can fuse or aggregate [54] [10]. Stabilization strategies include:
- PEGylation: Incorporating polyethylene glycol (PEG) lipids creates a steric barrier ("stealth" effect), reducing fusion and recognition by the immune system, thereby extending circulation time [9] [8].
- Composition Optimization: Adding cholesterol or using saturated lipids can increase bilayer rigidity and reduce drug leakage [10].
- Lyophilization: Freeze-drying is an effective method to extend the shelf-life of liposomes by removing water and preventing hydrolysis and aggregation during storage [9].
- Antioxidants: Including antioxidants like α-tocopherol in the formulation can scavenge free radicals and inhibit lipid oxidation [10].
Polymeric Nanoparticle Degradation and Stabilization
Polymeric nanoparticle instability centers on polymer degradation and aggregation. Polymer hydrolysis (via bulk or surface erosion) leads to molecular weight loss, matrix breakdown, and potentially unpredictable burst drug release [55] [22]. Colloidal aggregation occurs when nanoparticles aggregate in biological fluids due to salt or protein interactions, changing their distribution and uptake [55]. Stabilization strategies include:
- Stabilizers: Adding stabilizers like polyvinyl alcohol (PVA), Pluronic F-127, or Poloxamer 407 during formulation is critical for controlling particle size, achieving low PDI, and preventing aggregation [57].
- PEG Coating: Similar to liposomes, PEGylation provides a stealth coating, improving colloidal stability and circulation time [22].
- Polymer Selection: Choosing less hydrolytically sensitive polymers (e.g., PMMA) over others (e.g., PLA) for specific administration routes can prevent aggregation in certain biological environments [55].
- Pre-screening Stability: Conducting stability tests in various biological fluids (e.g., gastric juice, serum) ex vivo using DLS can predict in vivo aggregation and inform formulation design [55].
The Scientist's Toolkit: Key Reagents and Materials
Selecting the appropriate materials is fundamental to formulating stable nanocarriers. The following table lists essential components and their functions in mitigating instability.
Table 4: Essential Research Reagents for Stable Nano-Formulations
| Reagent/Material | Function in Formulation | Key Benefit/Role in Addressing Instability |
|---|---|---|
| DSPE-PEG (PEGylated lipid) [9] [8] | Stealth component incorporated into liposome bilayers. | Reduces opsonization and clearance (ABC phenomenon noted upon repeated dosing); enhances circulation time and physical stability against fusion. |
| Cholesterol [56] [10] | Additive integrated into lipid bilayers. | Increases membrane rigidity and stability, reducing drug leakage and improving vesicle physical integrity. |
| PLGA/PLA Polymers [11] [22] | Biodegradable matrix for polymeric nanoparticles. | Provides a controllable degradation profile for sustained drug release; degradation rate can be tuned by molecular weight and copolymer ratio. |
| Polyvinyl Alcohol (PVA) [57] | Stabilizer for polymeric nanoparticles. | Prevents nanoparticle aggregation during synthesis and storage, ensuring narrow size distribution (low PDI) and colloidal stability. |
| Pluronic F-127 / Poloxamer [57] | Amphiphilic stabilizer for PNPs. | Enhances physical stability of nanoparticles; can improve solubility of hydrophobic drugs and prevent burst release. |
| Antioxidants (e.g., α-Tocopherol) | Additive to lipid-based formulations. | Inhibits chemical degradation of phospholipids by scavenging free radicals, thereby preventing lipid oxidation. |
The choice between liposomal and polymeric nanoparticle formulations involves a careful trade-off between their inherent stability profiles. Liposomes offer superior biocompatibility and a biomimetic structure but require vigilant protection against chemical and physical degradation. Polymeric nanoparticles generally provide higher encapsulation efficiency and more robust, controllable release kinetics, but their stability is highly dependent on polymer selection and the use of effective stabilizers to prevent aggregation and manage degradation. The future of stable nanocarriers lies in advanced engineering strategies, such as the development of smart stimuli-responsive systems and hybrid nanoparticles, which combine the beneficial properties of both lipid and polymer systems to overcome these persistent instability challenges [54] [22] [8].
The selection of an appropriate nanocarrier system is a pivotal decision in drug development, influencing everything from initial therapeutic efficacy to final commercial viability. Among the most prominent candidates are polymeric nanoparticles, notably those made from poly(lactic-co-glycolic acid) (PLGA), and liposomal formulations. While both offer the fundamental advantages of nano-encapsulation—such as protecting active ingredients, controlling release profiles, and enhancing bioavailability—their inherent physicochemical differences lead to distinct challenges during scale-up. Polymeric nanoparticles are characterized by a solid, biodegradable polymer matrix that provides high structural integrity, whereas liposomes are microscopic vesicles composed of phospholipid bilayers enclosing an aqueous core [10]. This guide provides an objective, data-driven comparison of these two platforms, focusing on the critical scale-up challenges of batch-to-batch consistency, encapsulation efficiency, and cost-effective manufacturing, providing researchers with a clear framework for informed decision-making.
Comparative Performance: Polymeric Nanoparticles vs. Liposomal Formulations
A direct comparison of key performance metrics reveals fundamental differences between PLGA-based polymeric nanoparticles and liposomes, which become critically important when transitioning from laboratory synthesis to commercial-scale manufacturing.
Table 1: Direct Comparison of Key Formulation and Performance Metrics
| Parameter | Polymeric Nanoparticles (PLGA) | Liposomal Formulations | Experimental Context & Notes |
|---|---|---|---|
| Typical Particle Size | ~250 nm [11] | ~98 nm [11] | Model drug: L-Carnitine [11] |
| Zeta Potential | -32.8 mV [11] | +6.36 mV [11] | Model drug: L-Carnitine; varies with lipid composition [11] |
| Encapsulation Efficiency (EE) | 21.93% (L-Carnitine) [11]>87% (Meloxicam, SLN) [12] | 14.26% (L-Carnitine) [11] | Varies significantly with drug properties (hydrophilicity/lipophilicity) [11] [12] |
| Polydispersity Index (PDI) | 0.22 [11] | 0.35 [11] | Lower PDI indicates a more monodisperse, uniform population [11] |
| In Vitro Release Profile | Controlled release maintained for 12 hours [11] | Controlled release maintained for 12 hours [11] | Both showed similar sustained release profiles for L-Carnitine [11] |
| Structural Integrity | High (Rigid polymer matrix) [10] | Moderate (Dynamic lipid bilayer) [10] | Rigidity affects drug leakage and storage stability [10] |
Insights from Experimental Data
The data in Table 1, primarily derived from a comparative study of L-carnitine delivery systems, highlights several key trends. Polymeric nanoparticles (PLGA) in this instance demonstrated a higher encapsulation efficiency and a more uniform particle population (lower PDI) compared to their liposomal counterparts [11]. The significantly higher negative zeta potential of PLGA nanoparticles also suggests potentially greater colloidal stability due to stronger electrostatic repulsion between particles, which can be a critical factor during storage and shelf-life [11]. Furthermore, research on lipid-based Solid Lipid Nanoparticles (SLNs), which share characteristics with both systems, has shown encapsulation efficiency exceeding 87% for meloxicam, indicating that specific formulation choices can optimize this parameter [12].
Scale-Up Manufacturing: Methodologies and Industrial Translation
The journey from a benchtop formulation to a commercially viable product introduces a set of complex engineering challenges. The manufacturing process must be not only reproducible and scalable but also cost-effective.
Manufacturing Techniques and Workflows
The methodologies for producing nanoparticles at a laboratory scale differ significantly from those required for industrial manufacturing. Below is a generalized workflow comparing the scale-up paths for polymeric and liposomal formulations, highlighting critical control points.
Advanced and Cost-Effective Manufacturing Platforms
Innovative manufacturing platforms are crucial for overcoming traditional scale-up hurdles. A prominent advancement is the adoption of continuous flow methods over traditional batch processing.
- Microfluidic Synthesis: Technologies like T-mixer or cross-mixer systems allow for highly controlled mixing of organic and aqueous phases, leading to superior control over particle size and polydispersity [58]. This method is applicable to both liposomes and polymeric nanoparticles.
- Low-Cost, Accessible Platforms: Research demonstrates the successful repurposing of consumer-grade 3D printers into programmable syringe pumps for nanoparticle synthesis [58]. This setup, costing orders of magnitude less than commercial equipment, can synthesize liposomes, PLGA nanoparticles, and solid lipid nanoparticles by systematically controlling the aqueous-to-organic flow rate ratio (FRR) [58]. For liposomes, increasing the FRR from 3 to 15 results in a predictable decrease in hydrodynamic diameter and improved monodispersity (PDI <0.2 at FRRs ≥5) [58].
- Hybrid Methods: To merge the benefits of both systems, lipid-polymer hybrid nanoparticles have been developed. These typically feature a polymeric core for structural integrity and drug encapsulation, surrounded by a lipid coat that enhances biocompatibility and acts as a barrier to drug leakage [59]. These often require modified emulsification-solvent-evaporation methods [59].
Critical Analysis of Scale-Up Challenges
The transition to large-scale manufacturing amplifies specific challenges that may be manageable at the lab level but become critical for commercial success.
Table 2: Scale-Up Challenges and Mitigation Strategies
| Scale-Up Challenge | Impact on Polymeric Nanoparticles (PLGA) | Impact on Liposomal Formulations | Proven Mitigation Strategies |
|---|---|---|---|
| Batch-to-Batch Consistency | Challenge: Changes in reactor geometry & mixing dynamics alter reaction kinetics & heat transfer, risking product variability [60].Observation: PLGA NPs generally show low PDI, indicating good inherent consistency [11]. | Challenge: Achieving uniform lipid bilayer formation and vesicle size across large batches is difficult [61].Observation: Traditional methods can yield higher PDI; continuous flow improves this [58]. | - Implement Quality by Design (QbD) and Design of Experiments (DoE) [60].- Use continuous flow manufacturing over batch processing [58].- Employ advanced Process Analytical Technology (PAT) for real-time monitoring [62]. |
| Encapsulation Efficiency (EE) & Drug Leakage | Challenge: Poor encapsulation of water-soluble drugs due to fast drug leakage during emulsification [59]. | Challenge: For water-soluble drugs in the aqueous core, EE can be low, and drug is prone to leakage [9]. | - For polymers: Develop lipid-polymer hybrid nanoparticles to limit water-soluble drug transport [59].- For liposomes: Optimize lipid composition and add cholesterol to modify bilayer fluidity [61].- Use continuous methods, which can achieve near 100% EE for RNA in LNPs [58]. |
| Cost-Effective Manufacturing & Equipment | Challenge: Long lead times for custom reactors; material compatibility issues (e.g., moving from glass to stainless steel) [60]. | Challenge: Scaling traditional thin-film hydration is complex and costly; stability issues may require expensive lyophilization [9]. | - Partner with CDMOs with scalable equipment platforms [62] [63].- Use modular process skids for flexible, incremental scaling [60].- Adopt low-cost, in-house solutions like 3D-printed fluidic devices for prototyping and small-batch production [58]. |
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful formulation and scale-up depend on a foundational set of high-quality materials and reagents. The table below details key components for developing polymeric and lipid-based nanocarriers.
Table 3: Essential Reagents for Nanoparticle Formulation Research
| Reagent/Material | Function in Formulation | Common Examples & Notes |
|---|---|---|
| PLGA (Poly(lactic-co-glycolic acid)) | Biodegradable Polymer Core: Forms the structural matrix of the nanoparticle, controlling drug release kinetics through hydrolysis [11] [10]. | Available in varying lactic acid to glycolic acid ratios and molecular weights, which directly influence degradation rate and drug release profile. |
| Phospholipids | Bilayer Building Blocks: The primary structural component of liposomes, forming the amphiphilic bilayer [61] [10]. | Soy or egg lecithin, phosphatidylcholine, phosphatidylserine. Purified phosphatidylcholine is often used for entrapping bioactive peptides [61]. |
| Cholesterol | Membrane Modifier: Incorporated into lipid bilayers (liposomes/LNPs) to modify fluidity, enhance stability, and prevent crystallization of phospholipids [61] [9]. | Typically used at molar ratios of 1:1 or 2:1 (phospholipid:cholesterol) [61]. |
| PEGylated Lipids (Stealth Lipids) | Steric Stabilizer: Covalently attached PEG polymers create a hydrophilic "cloud" that reduces opsonization, prolonging circulation time (stealth effect) and improving physical stability [9]. | Examples: mPEG-DMG, ALC-0159, DSPE-PEG. Critical component in COVID-19 mRNA vaccines [9]. |
| Cationic Lipids | Nucleic Acid Delivery: Carry a positive charge to complex with and encapsulate negatively charged nucleic acids (DNA, mRNA) in lipid nanoparticles [9]. | Key for gene delivery and mRNA vaccines. Often used in combination with other lipid components. |
| PVA (Polyvinyl Alcohol) | Surfactant/Stabilizer: Commonly used in the emulsification step of PLGA nanoparticle preparation to prevent aggregation and control particle size [59]. | Often used in aqueous phase during single or double emulsification solvent evaporation methods. |
The choice between polymeric nanoparticles and liposomal formulations is not a matter of declaring a universal "best" option, but rather of aligning the strengths of each platform with a specific therapeutic and commercial goal. Polymeric nanoparticles (PLGA) often exhibit advantages in structural integrity, controlled release, and potentially higher encapsulation efficiency for certain hydrophobic compounds, making them a robust choice for sustained-release applications. Liposomal formulations, on the other hand, offer superior biocompatibility, high versatility in encapsulating both hydrophilic and hydrophobic agents, and have a proven track record in clinical translation, especially when combined with PEGylation for long circulation.
From a scale-up perspective, the emergence of continuous manufacturing platforms and advanced process controls is mitigating the traditional challenges of batch-to-batch consistency for both systems. The growing role of specialized Contract Development and Manufacturing Organizations (CDMOs) provides crucial expertise and infrastructure to navigate these complex scaling processes efficiently [62] [63]. Future research will continue to focus on overcoming encapsulation challenges for difficult drug molecules, optimizing green and cost-effective manufacturing, and developing even more sophisticated hybrid systems to harness the collective advantages of lipids and polymers for advanced drug delivery.
The efficacy of nanocarrier drug delivery systems (NDDS) is fundamentally hampered by innate biological clearance mechanisms following intravenous administration. The mononuclear phagocyte system (MPS), previously known as the reticuloendothelial system (RES), serves as the primary barrier, rapidly removing particles from circulation via phagocytic cells in the liver, spleen, and lymph nodes [64] [65]. To circumvent this, polyethylene glycol (PEG) coating became a cornerstone strategy, creating a steric "stealth" shield that reduces opsonin binding and MPS recognition, thereby prolonging circulation time [66] [64]. However, long-term efficacy of PEGylation is undermined by the accelerated blood clearance (ABC) phenomenon, where a second dose of PEGylated nanocarriers is cleared rapidly from the bloodstream, losing its long-circulating characteristics [67] [66]. This article provides a comparative analysis of polymeric nanoparticle and liposomal formulations, focusing on their interactions with these biological barriers and evaluating advanced strategies for effective immune evasion.
Understanding the Accelerated Blood Clearance (ABC) Phenomenon
Immunological Mechanism of the ABC Phenomenon
The ABC phenomenon represents a critical impediment to repeated dosing of nanocarriers. The widely accepted immunological mechanism involves a T cell-independent immune response orchestrated by the spleen [67] [64].
- Initial Exposure (Priming Dose): The first administration of PEGylated nanocarriers acts as a class-2 thymus-independent antigen (TI-2), stimulating specific B cells in the splenic marginal zone. This triggers the production and secretion of anti-PEG immunoglobulin M (IgM) antibodies [67] [64].
- Secondary Exposure (Eliciting Dose): Upon a repeated injection within a specific time window (days), pre-existing anti-PEG IgM antibodies instantly bind to the PEG chains on the nanocarrier surface. This binding forms an immune complex that activates the complement system via the classical pathway [67].
- Effector Phase: Complement activation results in the deposition of C3 opsonins on the nanocarrier surface. These opsonins are recognized by complement receptors on Kupffer cells (liver macrophages), leading to rapid phagocytosis and clearance of the second dose from the bloodstream, thus abrogating the stealth effect [67] [66].
The pivotal role of anti-PEG IgM has been demonstrated in studies where serum containing these antibodies was able to transfer the ABC phenomenon to naive animals [64]. Furthermore, splenectomized animals show a significant reduction in both anti-PEG IgM levels and the strength of the ABC phenomenon, confirming the spleen's central role [64].
The ABC Phenomenon Across Nanocarrier Types
The ABC phenomenon is not limited to a single nanocarrier type but has been documented across a spectrum of PEGylated formulations, though its intensity varies. A comparative study in beagle dogs investigated the immune response elicited by cross-administration of different nanocarriers and found the magnitude of the ABC phenomenon decreased in the following order: PEGylated liposomes (PL) > PEGylated emulsions (PE) > PEGylated solid lipid nanoparticles (PSLN) > PEG micelles (PM) [68]. This indicates that the colloidal structure and presentation of PEG (e.g., in a "brush" conformation on liposomes) significantly influence its immunogenicity [68]. Notably, the phenomenon can also be induced by repeated injections of some conventional, non-PEGylated liposomes, demonstrating that PEG modification, while a major trigger, is not an absolute prerequisite [64].
Comparative Analysis: Polymeric Nanoparticles vs. Liposomal Formulations
Fundamental Structural and Compositional Differences
Polymeric nanoparticles and liposomes represent two distinct classes of nanocarriers with unique structural attributes.
- Liposomes are spherical vesicles comprising one or more concentric phospholipid bilayers enclosing an aqueous interior, mimicking biological membranes. This structure allows for the encapsulation of hydrophilic drugs within the aqueous core and hydrophobic drugs within the lipid bilayer [10] [9]. They are typically sized between 50 and 200 nm [69].
- Polymeric Nanoparticles encompass a broader category, including solid matrices (e.g., PLGA), vesicles (polymersomes), and micelles. Polymersomes, analogous to liposomes, are formed from amphiphilic block copolymers that self-assemble into a bilayer vesicle with a hydrophilic core and a hydrophobic membrane. The higher molecular weight polymers confer a more compact and thicker membrane compared to liposomes [70]. Polymeric micelles, formed from diblock copolymers, possess a core-shell structure without an aqueous core, making them suitable primarily for hydrophobic drugs [69].
Table 1: Core Characteristics of Polymeric and Liposomal Nanocarriers
| Characteristic | Liposomes | Polymersomes | Polymeric Micelles |
|---|---|---|---|
| Structural Basis | Phospholipid bilayer | Amphiphilic block copolymer bilayer | Amphiphilic block copolymer core-shell |
| Typical Size Range | 50 - 200 nm [69] | 50 - 200 nm [70] | 10 - 100 nm [69] |
| Membrane Properties | Fluid, biologically similar | Thicker, more rigid, less permeable [70] | N/A (non-vesicular) |
| Drug Loading | Hydrophilic (core), Hydrophobic (bilayer) [10] | Hydrophilic (core), Hydrophobic (membrane) [70] | Primarily Hydrophobic (core) [69] |
| In Vivo Stability | Prone to drug leakage, fusion [70] [69] | High kinetic and physical stability [70] | High kinetic stability against dilution [69] |
Comparative Performance and Experimental Data
Direct comparative studies reveal how these structural differences translate into performance variations, particularly concerning stability, drug release, and immune evasion.
Table 2: Experimental Performance Comparison of Liposomes vs. Polymersomes Data adapted from a direct comparative study [70]
| Performance Metric | Liposomes (Egg PC/Chol) | Liposomes (Soybean PC/Chol) | Polymersome (P500) | Polymersome (P2000) |
|---|---|---|---|---|
| Hydrodynamic Radius (nm) | 135.0 | 141.2 | 125.6 | 138.5 |
| Polydispersity Index (PDI) | 0.18 | 0.22 | 0.21 | 0.18 |
| Zeta Potential (mV) | -1.0 | -3.2 | -3.8 | -5.5 |
| Encapsulation Efficiency (Hydrophilic Agent) | 4.3% | 5.8% | 9.4% | 11.2% |
| Encapsulation Efficiency (Hydrophobic Agent) | 74.1% | 78.9% | 86.5% | 88.2% |
| Stability (Size Increase after 30 days) | ~50% increase | ~25% increase | No significant change | No significant change |
Key Insights from Experimental Data:
- Superior Stability of Polymersomes: Polymersomes demonstrated exceptional physical stability over one month, with no significant change in size, whereas liposomes showed considerable aggregation and growth. This is attributed to the thicker, more rigid membrane of polymersomes and their lower critical aggregation concentration, which prevents dissociation upon dilution [70] [69].
- Enhanced Encapsulation Efficiency: Polymersomes consistently achieved higher encapsulation efficiencies for both hydrophilic and hydrophobic model compounds compared to their liposomal counterparts. This is likely due to their less permeable membrane, which better retains cargo during preparation and storage [70].
- Susceptibility to the ABC Phenomenon: While PEGylation extends the circulation half-life of both systems, it also makes them susceptible to the ABC phenomenon. The strength of the immune response can vary with the nanocarrier's core structure, with liposomes often eliciting a stronger response than other polymeric systems like micelles [68].
Advanced Strategies for Immune Evasion
Biomimetic Camouflage
A cutting-edge approach to circumvent both MPS clearance and the ABC phenomenon involves camouflaging nanocarriers with natural biological membranes.
- Cell Membrane Coating: Nanocarriers can be coated with membranes derived from red blood cells (RBCs), platelets, or leukocytes. This "cloaking" allows the particle to be recognized by the body as "self," thereby evading immune detection. RBC coating, for instance, can significantly reduce phagocytic uptake and extend circulation half-life [66] [65].
- "Hitchhiking" on Red Blood Cells: An alternative strategy involves the transient adsorption of nanocarriers onto the surface of RBCs. This hijacks the natural long-circulating properties of RBCs, preventing liver and spleen accumulation and enhancing delivery to target organs like the lungs [66].
MPS Blockade and Immunosuppression
This strategy involves temporarily inhibiting the phagocytic function of the MPS to create a window for nanocarrier delivery.
- Macrophage Depletion: Pre-administering macrophage-depleting agents like liposomal clodronate or gadolinium chloride can drastically reduce the population of Kupffer cells in the liver. This can enhance the tumor delivery of a subsequent nanocarrier dose by up to 150-fold [65]. However, the long-term immunosuppression and toxicity of these agents raise concerns for clinical translation.
- Complement Inhibition: Experimental approaches have used cobra venom factor (CVF) to deplete complement system components in vivo. Studies show that complement inhibition can weaken the ABC phenomenon and enhance the circulation time of a second PEGylated nanoemulsion dose, confirming the complement system's role in the effector phase. However, it does not completely abolish the phenomenon, indicating the involvement of other factors [67].
- Use of PEG Alternatives: To avoid anti-PEG immunity, researchers are exploring alternative stealth polymers, such as polysialic acid (PSA), polyglutamic acid (PGA), and poly(2-oxazoline)s, which offer stealth properties without eliciting a strong ABC response [66].
Experimental Protocols for Key Investigations
Objective: To evaluate the role of the complement system in the ABC phenomenon using cobra venom factor (CVF).
Methodology:
- Animal Model: Male Wistar rats (180-220 g).
- Complement Depletion: Administer CVF via injection to experimental group prior to the first nanocarrier dose. Control group receives a placebo.
- Nanocarrier Administration: Inject a priming dose of PEGylated nanoemulsion (PE) intravenously into both control and CVF-treated groups.
- Eliciting Dose: After a 7-day interval (to allow for anti-PEG IgM production), administer a second, identical dose of PE.
- Pharmacokinetic Analysis: Collect blood samples at various time points post-injection. Measure the blood concentration of a tracer drug (e.g., Tocopheryl Nicotinate) using HPLC.
- Data Analysis: Calculate the area under the blood concentration-time curve (AUC) for both the first and second doses. The ABC index is expressed as the ratio AUC
second/AUCfirst. A lower index indicates a stronger ABC phenomenon. Compare indices between CVF-treated and control groups. - Complement Activity Assay: Monitor total complement activity in serum using the CH50 hemolytic assay.
Objective: To directly compare the physical and drug retention stability of liposomes and polymersomes.
Methodology:
- Formulation Preparation:
- Liposomes: Prepare using thin-film hydration method with phosphatidylcholine and cholesterol.
- Polymersomes: Synthesize via self-assembly of amphiphilic block copolymers (e.g., PEG-polyester or PEG-polycarbonate).
- Characterization: Determine hydrodynamic diameter, polydispersity index (PDI), and zeta potential using dynamic light scattering.
- Encapsulation Efficiency: Load formulations with hydrophilic (e.g., FITC-dextran) and hydrophobic (e.g., anthracene) markers. Separate unencapsulated drug via dialysis or size exclusion chromatography. Calculate encapsulation efficiency (%) = (Amount of encapsulated drug / Total amount of drug used) * 100.
- In Vitro Drug Release: Place dialyzed formulations in phosphate-buffered saline (PBS) at 37°C under sink conditions. Collect samples at predetermined times and analyze drug content via HPLC or fluorescence spectroscopy. Plot cumulative drug release over time.
- Long-term Stability: Store formulations at 4°C and room temperature. Monitor changes in particle size and PDI over 30 days. A significant increase indicates aggregation and poor stability.
Visualizing Key Concepts and Workflows
The Mechanism and Investigation of the ABC Phenomenon
Diagram 1: Mechanism of the ABC phenomenon, illustrating the immunological cascade from initial exposure to rapid clearance of the second dose.
Experimental Workflow for MPS Blockade Strategy
Diagram 2: Workflow for MPS blockade strategy, showing pre-administration of blockers to saturate or deplete macrophages, enabling improved delivery of therapeutic nanocarriers.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Investigating Nanocarrier Clearance and Immune Evasion
| Reagent / Material | Function / Application | Specific Examples |
|---|---|---|
| PEGylated Lipids/Polymers | Imparts "stealth" properties to nanocarriers; subject of ABC studies. | mPEG2000-DSPE [67] [68], PEG-methacrylate [70] |
| Cobra Venit Factor (CVF) | Tool for in vivo complement system depletion to study its role in ABC [67]. | Isolated from snake venom [67] |
| Liposomal Clodronate | Agent for selective depletion of phagocytic MPS cells [65]. | Commercially available liposomal formulation |
| Phospholipids | Primary structural component of liposomes. | L-α-Phosphatidylcholine (from egg or soybean) [70], Soy Lecithin (S75) [67] |
| Amphiphilic Block Copolymers | Structural basis for self-assembled polymeric nanocarriers. | PEG-PLGA, PEG-PCL, custom-synthesized polymers [70] |
| CH50 Assay Kit | Measures total hemolytic complement activity in serum [67]. | Commercial kits based on sheep red blood cell lysis |
| Anti-PEG IgM/IgG ELISA Kit | Quantifies anti-PEG antibody levels in serum, critical for ABC studies [64]. | Commercial kits available |
The strategic choice between polymeric nanoparticles and liposomal formulations is multifaceted, requiring a careful balance of stability, drug loading, and immunological considerations. Polymersomes offer distinct advantages in physical stability and encapsulation efficiency, while liposomes have a more established clinical track record. However, both systems are susceptible to the biological barriers of MPS clearance and the ABC phenomenon. Future success in nanocarrier drug delivery hinges on the intelligent design of advanced biomimetic systems and the sophisticated temporal use of immunomodulatory strategies. Moving beyond pure PEGylation towards these next-generation approaches is paramount for realizing the full clinical potential of nanomedicine.
In the development of modern nanomedicines, particularly when comparing advanced delivery systems like polymeric nanoparticles and liposomes, rigorous physicochemical characterization is paramount. These parameters directly dictate the in vivo behavior, including stability, biodistribution, cellular uptake, and therapeutic efficacy of the nanocarriers [71]. For researchers and drug development professionals, a deep understanding of Critical Quality Attributes (CQAs) such as particle size, size distribution (polydispersity), zeta potential, and drug release profile is non-negotiable for ensuring product quality, batch-to-batch consistency, and successful regulatory approval [72] [73]. This guide provides a comparative analysis of these essential techniques, framed within the context of polymeric nanoparticle and liposomal formulation research, offering structured experimental data and detailed protocols to inform development workflows.
Critical Quality Attributes (CQAs) in Nanomedicine
The performance of a nanocarrier is a direct function of its physicochemical properties. Particle size and size distribution are perhaps the most fundamental CQAs. They influence circulation time, cellular uptake mechanisms, and the Enhanced Permeability and Retention (EPR) effect in oncology applications. Nanoparticles intended for intravenous injection and passive tumor targeting typically require a size range of 10 to 200 nm to leverage the EPR effect effectively, as larger particles are more prone to phagocytosis by macrophages [71]. The polydispersity index (PDI) is a key metric for homogeneity, where a value below 0.3 is generally indicative of a monodisperse, stable formulation [11] [74].
The zeta potential, which reflects the surface charge of nanoparticles in a colloidal suspension, is a critical indicator of physical stability. It measures the electrostatic potential at the hydrodynamic slipping plane surrounding the particle. High absolute zeta potential values (generally ≥ ±30 mV) signify strong electrostatic repulsion between particles, preventing aggregation and ensuring long-term stability. Conversely, formulations with a low zeta potential (e.g., ≤ ±10 mV) are prone to coagulation and flocculation [71] [72]. Furthermore, zeta potential significantly impacts biological interactions, including cellular uptake and protein adsorption [72].
Finally, the drug release profile is a functional CQA that verifies the nanocarrier's ability to modulate the release of its payload. A controlled, sustained release profile is often a primary objective, aiming to maintain therapeutic drug levels over an extended period and reduce dosing frequency compared to conventional formulations [11] [74].
Comparative Analysis of Formulation CQAs
The table below summarizes typical CQA data for a model compound (L-carnitine) formulated in both a liposomal and a polymeric (PLGA) nanoparticle system, illustrating the distinct physicochemical profiles of these two carrier types [11] [74].
Table 1: Comparative Physicochemical Properties of Liposomal and PLGA Nanoparticle Formulations of L-Carnitine
| Critical Quality Attribute (CQA) | Liposomal Formulation (Lipo-Carnitine) | PLGA Nanoparticle Formulation (Nano-Carnitine) |
|---|---|---|
| Particle Size (nm) | 97.88 ± 2.96 nm | 250.90 ± 6.15 nm |
| Polydispersity Index (PDI) | 0.35 ± 0.01 | 0.22 ± 0.03 |
| Zeta Potential (mV) | +6.36 ± 0.54 mV | -32.80 ± 2.26 mV |
| Encapsulation Efficiency (%) | 14.26 ± 3.52% | 21.93 ± 4.17% |
| Drug Release Duration | Sustained release over 12 hours | Sustained release over 12 hours |
Experimental Protocols for CQA Assessment
Particle Size and Polydispersity Index (PDI) Analysis
Principle: Dynamic Light Scattering (DLS), also known as Photon Correlation Spectroscopy, is the standard technique for determining the hydrodynamic diameter and size distribution of nanoparticles in suspension. It analyzes the fluctuations in the intensity of scattered light caused by Brownian motion of particles.
Methodology:
- Sample Preparation: Dilute the nanoparticle dispersion (liposomal or polymeric) with an appropriate filtered buffer (e.g., 1 mM KCl or purified water) to achieve a faintly opalescent solution, ensuring the scattering intensity is within the instrument's optimal range.
- Instrumentation: Use a Zetasizer Nano or equivalent DLS instrument.
- Measurement: Transfer the diluted sample into a disposable sizing cuvette. The measurement temperature should be equilibrated (typically 25°C). Perform a minimum of three measurements per sample.
- Data Analysis: The instrument software calculates the intensity-weighted mean particle diameter (Z-average) and the Polydispersity Index (PDI). Report the Z-average diameter and PDI as mean ± standard deviation from the replicates. A PDI value below 0.3 is typically associated with a monodisperse population [11] [74].
Zeta Potential Analysis
Principle: Zeta potential is measured using Laser Doppler Velocimetry. An electrical field is applied to the nanoparticle suspension, causing charged particles to move towards the oppositely charged electrode. Their velocity (electrophoretic mobility) is measured and converted to zeta potential using the Henry equation.
Methodology:
- Sample Preparation: Similar to size analysis, dilute the sample with a filtered, low-conductivity buffer. The use of purified water is acceptable if the particles remain stable, but a standard saline solution like 1 mM KCl can be used to control ionic strength.
- Instrumentation: Use the same Zetasizer Nano instrument with a dedicated zeta potential dip cell.
- Measurement: Load the sample into a clear, disposable zeta cell, ensuring no air bubbles are present. Set the instrument parameters (temperature, field strength) and perform a minimum of 3-12 runs per measurement.
- Data Analysis: The software reports the zeta potential in millivolts (mV). Report the mean value and standard deviation. As per the literature, formulations with a zeta potential ≥ ±30 mV are considered physically stable due to strong electrostatic repulsion [71] [72].
In Vitro Drug Release Profiling
Principle: This assay evaluates the release kinetics of the encapsulated drug from the nanocarrier under simulated physiological conditions. The dialysis method is most commonly employed.
Methodology:
- Setup: Place a precise volume of the nanoparticle formulation (e.g., equivalent to 1 mg of drug) into a pre-soaked dialysis membrane tube (with an appropriate molecular weight cut-off, e.g., 12-14 kDa).
- Release Medium: Immerse the sealed dialysis bag in a large volume (e.g., 200-500 mL) of release medium (e.g., Phosphate Buffered Saline, PBS, at pH 7.4) maintained at 37°C under constant, gentle agitation. The large volume ensures "sink conditions" are maintained.
- Sampling: At predetermined time intervals (e.g., 0.5, 1, 2, 4, 8, 12, 24 hours), withdraw a small aliquot (e.g., 1 mL) from the release medium and replace it with an equal volume of fresh, pre-warmed medium to maintain the sink condition.
- Analysis: Quantify the drug concentration in the withdrawn samples using a validated analytical method, such as High-Performance Liquid Chromatography (HPLC) with UV detection.
- Data Analysis: Calculate the cumulative percentage of drug released and plot it against time to generate the release profile. Compare the profile of the nano-formulation (e.g., sustained release over 12 hours) with that of a pure drug solution (e.g., 90% release within 1 hour) to demonstrate the controlled-release capability [11] [74].
Figure 1: Workflow for essential nanoparticle characterization techniques.
The Scientist's Toolkit: Key Reagents and Materials
Successful characterization relies on high-quality reagents and materials. The following table lists essential solutions and their specific functions in the characterization process.
Table 2: Essential Research Reagent Solutions for Nanoparticle Characterization
| Research Reagent Solution | Function / Rationale |
|---|---|
| Phosphate Buffered Saline (PBS) | A standard isotonic buffer used as a dilution medium for size and zeta potential measurements, and as a common release medium for in vitro drug release studies at physiological pH [11]. |
| Filtered 1 mM Potassium Chloride (KCl) | A low-conductivity solution ideal for zeta potential measurements, as high salt concentrations can compress the electrical double layer and mask the true surface potential. Must be filtered through a 0.2 µm or 0.1 µm membrane before use. |
| Dialysis Membranes (e.g., MWCO 12-14 kDa) | Semi-permeable tubes used in drug release studies to separate the nanoparticles from the release medium, allowing only the free drug to diffuse out. The Molecular Weight Cut-Off (MWCO) must be selected to retain the nanocarrier. |
| Poloxamer 407 / Tween 80 | Common surfactants and anti-solvents used in the preparation and stabilization of polymeric nanoparticles like PLGA, preventing aggregation during and after synthesis [75]. |
| Stearylamine (SA) / Dicetyl Phosphate (DCP) | Charge-imparting agents added to liposomal formulations during the thin-film hydration process to modify the surface charge (zeta potential). SA confers a positive charge, while DCP confers a negative charge [72]. |
| Polyethylene Glycol (PEG) Lipids | Used in the preparation of "stealth" or PEGylated liposomes. PEGylation creates a hydrophilic layer on the liposome surface, prolonging circulation time by reducing opsonization and uptake by the mononuclear phagocyte system [8]. |
Advanced Considerations and Quality Control
Optimizing and Controlling Zeta Potential
The surface charge of nanocarriers is not a fixed property but can be rationally designed. For liposomes, the zeta potential can be precisely modulated by incorporating charge-imparting additives into the lipid bilayer. As demonstrated in a Quality by Design (QbD) study:
- Stearylamine (SA) is used to create a positive surface charge. An optimized molar ratio of phosphatidylcholine:cholesterol:SA of 12.0:5.0:5.0 can yield liposomes with a zeta potential of +30.1 ± 1.2 mV [72].
- Dicetyl Phosphate (DCP) is used to create a stronger negative surface charge. An optimized molar ratio of 8.5:4.5:6.5 can produce liposomes with a zeta potential of -36.7 ± 3.3 mV [72].
For polymeric nanoparticles, machine learning (ML) models are emerging as powerful tools for predicting and optimizing CQAs like size and zeta potential based on synthesis input parameters (e.g., polymer type, concentration, anti-solvent type). Gaussian Process Regression (GPR) has shown exceptional performance in predicting these attributes, which can significantly reduce experimental time and cost [75].
Stability and Biological Performance
Zeta potential is a key predictor of a formulation's colloidal stability during storage. Furthermore, it significantly influences in vivo behavior. Positively charged nanoparticles typically exhibit stronger binding to negatively charged cell membranes, leading to higher cellular uptake [72]. However, for mucus-rich administration routes (e.g., nasal, pulmonary), neutral or negatively charged nanoparticles demonstrate better diffusion through the negatively charged mucus network, while positively charged particles tend to be immobilized [71].
Figure 2: Impact of zeta potential on stability and biological performance.
The direct comparison between liposomal and PLGA nanoparticle formulations for L-carnitine delivery underscores a critical principle in nanomedicine: the choice of carrier system produces a distinct physicochemical fingerprint, each with its own advantages and challenges. Liposomes offered a smaller particle size, while the PLGA nanoparticles provided a higher negative zeta potential and superior encapsulation efficiency in this specific study [11] [74]. This highlights that there is no universally "superior" system; the optimal choice depends entirely on the therapeutic molecule, the target tissue, and the desired release profile.
Mastering the techniques of size, zeta potential, and drug release profiling is not merely a regulatory checkbox but a fundamental practice in rational nanocarrier design. By employing the detailed experimental protocols outlined in this guide and leveraging advanced strategies such as QbD for formulation optimization and machine learning for predictive synthesis, scientists can systematically develop robust, efficacious, and high-quality nanomedicines. The future of the field lies in this deep, data-driven understanding and precise control over these critical quality attributes.
Head-to-Head Evaluation: Clinical Efficacy, Commercial Success, and Decision Matrices
In the evolving landscape of nanomedicine, polymeric nanoparticles (PNPs) and liposomal formulations represent two of the most prominent platforms for targeted drug delivery. Within the broader context of ongoing pharmaceutical research, a critical and objective comparison of their fundamental properties—drug loading capacity, release kinetics, and storage stability—is essential for rational carrier selection and development. This guide provides a side-by-side examination of these key parameters, underpinned by experimental data and methodologies, to inform researchers and drug development professionals.
Comparative Analysis of Key Properties
The following tables provide a direct, data-driven comparison of the core properties of polymeric and liposomal nanoparticles, summarizing findings from recent scientific literature.
Table 1: Comparative Drug Loading Capacity and Release Kinetics
| Property | Polymeric Nanoparticles (PNPs) | Liposomal Formulations |
|---|---|---|
| Drug Loading Capacity | High drug-loading capacity due to versatile polymer matrices and chemical functionalization [22] [24]. | Good encapsulation, enhanced by active loading techniques (e.g., ion gradients) and optimized lipid composition [23]. |
| Encapsulation Efficiency | Capable of high encapsulation efficiency for both hydrophilic and hydrophobic agents [24]. | Efficiency depends on drug properties and method; passive/active loading can achieve high efficiency [23]. |
| Influence of Composition | Polymer characteristics (MW, hydrophobicity) directly influence loading capacity [22]. | Lipid composition, cholesterol content, and charge critically determine drug loading [23] [76]. |
| Release Kinetics Profile | Sustained and controlled release, tunable from days to weeks [22] [77]. | Biphasic release (initial burst followed by slower release); can be modified for sustained release [8]. |
| Control Mechanisms | Engineered for precise control via polymer degradation and stimuli-responsive triggers (pH, enzymes) [22] [1]. | Controlled by bilayer composition, PEGylation, and integration of stimuli-responsive lipids [23] [8]. |
Table 2: Comparative Storage Stability and Clinical Translation
| Property | Polymeric Nanoparticles (PNPs) | Liposomal Formulations |
|---|---|---|
| Physical Stability | Generally superior physical stability, resisting fusion and aggregation [1]. | Prone to physical instability: drug leakage, fusion, and aggregation over time [23]. |
| Chemical Stability | Good chemical stability of polymer matrices [24]. | Susceptible to chemical degradation: lipid oxidation and hydrolysis [23]. |
| Storage Considerations | Often stored as stable aqueous dispersions or lyophilized powders [1]. | Lyophilization with cryoprotectants often required for long-term storage [23]. |
| Circulation Stability | PEGylation and surface functionalization enhance circulation time and stability in blood [22]. | PEGylation extends circulation but can induce Accelerated Blood Clearance (ABC) phenomenon upon repeated dosing [23] [8]. |
| Scalability & Reproducibility | Challenges in batch-to-batch reproducibility and scalable manufacturing of complex polymers [22]. | Well-established large-scale production, though batch uniformity and sterilization remain challenging [23]. |
Experimental Protocols for Key Characterization
Robust experimental characterization is fundamental for comparing nanoparticle properties. Below are detailed methodologies for assessing the key parameters discussed.
Protocol for Determining Drug Loading Capacity and Encapsulation Efficiency
This protocol is applicable to both PNPs and liposomes to quantify the amount of drug successfully incorporated into the nanocarrier.
- Principle: Separation of unencapsulated/free drug from the nanoparticle formulation, followed by quantification of the drug in each fraction.
- Materials: Drug-loaded nanoparticles, appropriate buffer, centrifugal filters (e.g., 10-100 kDa MWCO) or size-exclusion chromatography columns (e.g., Sephadex G-50), analytical instrument for drug quantification (e.g., HPLC, UV-Vis spectrophotometer).
- Method Steps:
- Purification: A known volume of the nanoparticle suspension is purified using a centrifugal filter device by centrifuging at a predetermined force and time to separate the free drug (in the filtrate) from the drug-loaded nanoparticles (in the retentate). Alternatively, size-exclusion chromatography can be used.
- Lysis of Nanoparticles: The purified nanoparticle fraction (retentate) is lysed using a suitable solvent (e.g., organic solvent like acetonitrile for PNPs, or detergent like 1% Triton X-100 for liposomes [76]) to release the encapsulated drug.
- Quantification:
- Encapsulated Drug: The lysed nanoparticle solution is analyzed to determine the concentration of the encapsulated drug (Cencapsulated).
- Free Drug: The filtrate from step 1 is analyzed to determine the concentration of the free, unencapsulated drug (Cfree).
- Total Drug: A separate sample of the initial, unpurified nanoparticle suspension is directly lysed and analyzed to determine the total drug concentration (C_total).
- Calculations:
- Drug Loading Capacity (DLC): (Mass of encapsulated drug / Mass of nanoparticles) × 100%.
- Encapsulation Efficiency (EE): (Mass of encapsulated drug / Total mass of drug) × 100% = (Cencapsulated / Ctotal) × 100%.
Protocol for Assessing In Vitro Drug Release Kinetics
This protocol determines the rate at which the drug is released from the nanoparticle under controlled conditions, simulating the biological environment.
- Principle: Incubating the drug-loaded nanoparticles in a release medium and measuring the amount of drug released over time.
- Materials: Drug-loaded nanoparticles, release medium (e.g., PBS at pH 7.4, or acetate buffer at pH 5.5 to simulate endosomes), dialysis membrane bags (with appropriate MWCO) or a continuous flow system, sampling vessel, analytical instrument (e.g., HPLC, UV-Vis).
- Method Steps (Dialysis):
- Setup: A precise volume of the nanoparticle formulation is placed in a dialysis bag, which is securely sealed.
- Incubation: The bag is immersed in a large volume of release medium (sink condition) and maintained at a constant temperature (e.g., 37°C) with continuous agitation.
- Sampling: At predetermined time intervals, small aliquots of the external release medium are withdrawn for analysis. An equal volume of fresh pre-warmed release medium is immediately replaced to maintain sink conditions.
- Analysis: The concentration of the drug in each sample is quantified using a calibrated analytical method.
- Data Processing: The cumulative percentage of drug released is plotted against time to generate the release profile and determine the release kinetics [76].
- Advanced Methods: For complex media or intracellular release assessment, alternative methods like flow cytometry with fluorescent model drugs (e.g., sulforhodamine B) can be employed [76].
Protocol for Evaluating Storage Stability
This protocol assesses the physical and chemical integrity of nanoparticle formulations over time under defined storage conditions.
- Principle: Monitoring changes in critical physicochemical parameters of nanoparticles during storage.
- Materials: Nanoparticle formulation, storage vials, controlled temperature incubators (e.g., 4°C, 25°C), analytical instruments: Dynamic Light Scattering (DLS) for size, Zeta Potential Analyzer for surface charge, HPLC for drug content and degradation products.
- Method Steps:
- Study Design: Aliquots of the nanoparticle formulation are stored under various conditions (e.g., 4°C, 25°C/60% relative humidity, and possibly 40°C/75% RH for accelerated studies). Lyophilized samples may also be tested.
- Sampling: Samples are withdrawn at predefined time points (e.g., 0, 1, 3, 6 months).
- Analysis: At each time point, samples are analyzed for:
- Particle Size and PDI: Using DLS to detect aggregation or size change [1].
- Zeta Potential: To monitor surface charge changes, which can indicate instability.
- Drug Content: Using HPLC to determine the percentage of remaining active drug.
- Visual Inspection: For precipitate formation or color change.
- Chemical Stability: For liposomes, specific tests for phospholipid hydrolysis or oxidation (e.g., using thiobarbituric acid reactive substances (TBARS) assay) may be conducted [23].
Visualization of Properties and Workflows
The following diagrams illustrate the core concepts and experimental workflows discussed in this guide.
Drug Loading and Release Mechanisms
Stability Challenges and Solutions
The Scientist's Toolkit: Essential Research Reagents
This table lists key reagents and materials essential for the formulation and characterization of polymeric and liposomal nanoparticles, as referenced in the experimental protocols.
Table 3: Key Research Reagent Solutions for Nanoparticle Development
| Reagent/Material | Function/Application | Specific Examples & Notes |
|---|---|---|
| Biodegradable Polymers | Forms the core matrix of PNPs for drug encapsulation and controlled release. | Poly(lactic-co-glycolic acid) (PLGA), Polycaprolactone (PCL), Chitosan [22] [24]. |
| Phospholipids & Cholesterol | Fundamental building blocks of liposomal bilayers; cholesterol enhances membrane stability. | 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), DOPG; Cholesterol reduces drug leakage [23] [76]. |
| PEGylated Lipids/Polymers | Imparts "stealth" properties by reducing opsonization and extending circulation half-life. | DSPE-PEG, PEG-PLGA; note potential for Accelerated Blood Clearance (ABC) [23] [8]. |
| Ammonium Sulfate Solution | Used to create transmembrane pH gradients for active loading of weak base drugs into liposomes. | Critical for achieving high drug loading in liposomes (e.g., in Doxil) [23]. |
| Cryoprotectants | Prevents nanoparticle aggregation and fusion during lyophilization (freeze-drying). | Sugars (sucrose, trehalose) or polymers (PVA) [23]. |
| Detergents | Used to lyse liposomes and PNPs in release studies and encapsulation efficiency assays. | Triton X-100 [76]. |
| Analytical Standards | Essential for calibrating instruments and quantifying drug content, size, and charge. | Drug standards for HPLC, size standards for DLS, zeta potential standards [1]. |
The evolution of nanomedicine has positioned lipid- and polymer-based nanoparticles as cornerstone technologies for advanced drug delivery. Among these, liposomes and polymeric nanoparticles (PNPs) are the most extensively investigated, each offering distinct mechanisms to enhance drug solubility, prolong circulation, facilitate targeted delivery, and improve therapeutic outcomes. [78] Liposomes, with their biocompatible lipid bilayers, have transitioned from a laboratory concept to clinical reality, with formulations like Doxil demonstrating proven success in oncology. [23] [9] Concurrently, PNPs, particularly those made from biodegradable poly(lactic-co-glycolic acid) (PLGA) and polycaprolactone (PCL), offer exceptional tunability of drug release profiles and polymer chemistry, making them a versatile "laboratory" for nanomedicine design. [78] [79] However, the translational pathway from preclinical promise to clinical application is fraught with challenges, including biological barriers, manufacturing complexities, and regulatory hurdles. [78] This guide provides a objective, data-driven comparison of liposomal and polymeric nanoparticle formulations, dissecting their performance across key disease areas to inform researchers and drug development professionals.
Comparative Formulation Properties and Methodologies
The fundamental differences in the composition and structure of liposomes and polymeric nanoparticles directly influence their physicochemical properties, loading capacity, and the methodologies used for their preparation and drug encapsulation.
Physicochemical Properties and Loading Capacity
A direct comparative study of L-carnitine-loaded formulations highlights the distinct property profiles of these two systems (Table 1). [11]
Table 1: Comparative Physicochemical Properties of L-Carnitine-Loaded Formulations
| Parameter | Liposome (lipo-carnitine) | PLGA Nanoparticle (nano-carnitine) |
|---|---|---|
| Average Particle Size (nm) | 97.88 ± 2.96 nm | 250.90 ± 6.15 nm |
| Polydispersity Index (PDI) | 0.35 ± 0.01 | 0.22 ± 0.03 |
| Zeta Potential (mV) | 6.36 ± 0.54 mV | -32.80 ± 2.26 mV |
| Encapsulation Efficiency (%) | 14.26 ± 3.52% | 21.93 ± 4.17% |
The data shows that the liposomal formulation achieved a smaller particle size, which can be advantageous for tissue penetration. In contrast, the PNP exhibited a more homogeneous size distribution (lower PDI), a highly negative surface charge, and a superior encapsulation efficiency for the hydrophilic drug L-carnitine. [11] The negative zeta potential of PLGA nanoparticles contributes to their colloidal stability by promoting electrostatic repulsion between particles. Beyond this specific case, PNP systems, including PCL nanospheres for antibiotic delivery, have demonstrated encapsulation efficiencies as high as 84.5%. [79]
Key Experimental Protocols and Workflows
The preparation of these nanocarriers relies on distinct protocols tailored to their chemical nature.
Liposome Preparation (Thin-Film Hydration & Active Loading): A common and versatile method for liposome production is thin-film hydration. [80] This process involves (1) dissolving phospholipids and cholesterol in an organic solvent; (2) evaporating the solvent to form a thin lipid film; (3) hydrating the film with an aqueous buffer to form multilamellar vesicles; and (4) downsizing the vesicles via extrusion or sonication to obtain unilamellar liposomes of a defined size. [80] Drug loading can be passive (during hydration) or active. Active loading, or remote loading, is a powerful technique to achieve high drug encapsulation. It involves creating a transmembrane gradient (e.g., pH or ammonium sulfate) after the blank liposomes are formed. [23] [80] Weakly basic or acidic drug molecules can then diffuse across the membrane and become trapped in the ionized form within the liposome's aqueous interior, achieving concentrations up to 250 mM. [80]
Polymeric Nanoparticle Preparation (Double Emulsion Solvent Evaporation): For encapsulating hydrophilic drugs or biologics, the double emulsion (w/o/w) solvent evaporation method is widely used for PNPs. The protocol for Imipenem-loaded PCL nanospheres exemplifies this process (Figure 1). [79]
Figure 1: Experimental workflow for preparing drug-loaded polymeric nanoparticles using the double emulsion method. [79]
The process involves (1) emulsifying an inner aqueous drug solution into a polymer-containing organic phase (e.g., dichloromethane) to form a primary water-in-oil (w/o) emulsion; (2) this primary emulsion is then emulsified into a second aqueous phase containing a stabilizer like polyvinyl alcohol (PVA) to form a double (w/o/w) emulsion; (3) the organic solvent is evaporated by stirring, leading to the hardening of the polymer and the formation of solid nanospheres; and (4) the nanoparticles are collected by centrifugation, washed, and often lyophilized for storage. [79] This method is particularly effective for protecting water-soluble biologics and drugs from harsh physiological environments. [81]
Comparative Preclinical and Clinical Performance
The efficacy of liposomes and PNPs has been evaluated across various disease models, from cancer to infectious diseases, revealing distinct strengths and therapeutic profiles.
Performance in Oncology
Liposomes have established a strong foothold in cancer therapy, with their performance extensively documented. A key advantage is their ability to leverage the Enhanced Permeability and Retention (EPR) effect for passive tumor targeting. [80] In breast cancer, for instance, liposomes functionalized with targeting ligands (e.g., against HER2 receptors) can actively bind to cancer cells, enhancing cellular uptake and specificity. [80] Doxil, a PEGylated liposomal doxorubicin, demonstrates the clinical success of this platform, showing significantly prolonged circulation and reduced cardiotoxicity compared to free doxorubicin. [78] However, the clinical translation of this EPR-based passive targeting has been challenging, as the effect is highly heterogeneous and less pronounced in human patients than in animal models. [78]
Polymeric Nanoparticles are emerging as a highly tunable alternative, especially for controlled release and overcoming drug resistance. Smart PNPs (SPNs) can be engineered to respond to specific tumor microenvironment (TME) cues such as low pH, hypoxia, or specific enzymes, enabling localized drug release. [82] In aggressive cancers like pancreatic ductal adenocarcinoma (PDAC), SPNs are being designed to not only target cancer cells but also to actively remodel the dense fibrotic stroma or deliver matrix-modulating agents to improve drug penetration. [82] Formulations like Genexol-PM (polymeric micelles of paclitaxel) and Abraxane (albumin-bound paclitaxel) highlight the clinical relevance of polymer-based systems, offering improved drug solubility and tumor accumulation. [83] Recent studies report that advanced PNPs can achieve drug loading efficiencies of 80-90%, extend circulation half-lives by 2-5 fold, and improve tumor accumulation by 3-10 times compared to free drugs. [83]
Performance in Infectious Diseases
Nanoparticle platforms have shown significant promise in revitalizing the efficacy of antibiotics against resistant pathogens.
A compelling case study involves combating Carbapenem-Resistant Klebsiella pneumoniae (CRKP). Research demonstrates that encapsulating imipenem within polycaprolactone (PCL) nanospheres dramatically enhances its antibacterial potency (Table 2). [79]
Table 2: Efficacy of Imipenem-Loaded PCL Nanospheres against CRKP [79]
| Parameter | Free Imipenem | Imipenem-Loaded PCL Nanospheres |
|---|---|---|
| Minimum Inhibitory Concentration (MIC) | Baseline | 8-fold reduction |
| Biofilm Formation | Not Reported | Significantly Inhibited |
| Bacterial Killing Rate | Standard | Accelerated |
| Cytocompatibility (Fibroblasts) | Not Reported | >80% Cell Viability |
The PCL-based delivery system provided a sustained release profile, inhibited biofilm formation, and suppressed the expression of key bacterial resistance genes (blaKPC, blaNDM). [79] This multifaceted approach, combining enhanced drug delivery with resistance modulation, underscores the potential of PNPs to address complex antimicrobial resistance mechanisms.
Liposomes also offer strategic advantages for managing infections. Their structure allows for the co-encapsulation of both hydrophilic and hydrophobic drugs, enabling combination therapy. [23] Furthermore, strategies like PEGylation ("stealth" liposomes) and surface functionalization with ligands can prolong systemic circulation and direct liposomes to specific infection sites. [23] [9] Stimuli-responsive liposomes that release their payload in response to the slightly acidic pH or specific enzymes at infection sites are also under development to further improve specificity and efficacy. [23]
Clinical Translation and Pathway Analysis
Despite the wealth of preclinical research, the journey of nanomedicines from the laboratory to the clinic is marked by a significant "translational gap." [78] While over 100,000 scientific articles on nanomedicine have been published, only an estimated 90 nanomedicine products had gained global marketing approval as of 2023, with the portfolio dominated by liposomes, nanocrystals, and lipid nanoparticles. [78]
The clinical translation pathway for any nanomedicine is complex and requires careful navigation of multiple barriers beyond efficacy (Figure 2).
Figure 2: Key hurdles in the clinical translation of nanomedicines. [78]
Liposomes currently have a more mature regulatory track record, with several approved products. Their primary challenges in translation include potential immune reactions (e.g., the "accelerated blood clearance" phenomenon upon repeated dosing of PEGylated liposomes), physical instability leading to drug leakage, and complexities in large-scale manufacturing and sterilization. [78] [23]
Polymeric Nanoparticles face steeper translational hurdles, primarily related to Chemistry, Manufacturing, and Controls (CMC). The chemical diversity of polymers, while a design strength, often leads to challenges in achieving batch-to-batch reproducibility and scaling up under Good Manufacturing Practice (GMP) standards. [78] Furthermore, ensuring complete biocompatibility and safe biodegradation of novel synthetic polymers, while avoiding biopersistence and toxicity, is a critical regulatory concern. [78] The failure of targeted PNPs like BIND-014 in Phase II trials, despite promising preclinical data, highlights the difficulty in predicting clinical performance and the limitations of an over-reliance on the EPR effect. [78]
The Scientist's Toolkit: Essential Research Reagents
The development and evaluation of liposomal and polymeric nanoparticle formulations rely on a core set of materials and analytical techniques. The following table details key reagents and their functions in nanoparticle research.
Table 3: Essential Research Reagents and Materials for Nanoparticle Formulation
| Reagent/Material | Function in Formulation | Common Examples |
|---|---|---|
| Phospholipids | Fundamental building blocks of liposome bilayers. Provide amphiphilic structure. | Phosphatidylcholine (PC), saturated/unsaturated lipids. [23] [80] |
| Cholesterol | Incorporated into lipid bilayers to enhance membrane integrity and rigidity, reducing premature drug leakage. [23] | Plant-derived or synthetic cholesterol. [9] |
| Biodegradable Polymers | Form the matrix of polymeric nanoparticles, controlling degradation and drug release kinetics. | PLGA, Polycaprolactone (PCL). [11] [79] |
| PEGylated Lipids/Polymers | Impart "stealth" properties by creating a hydrophilic corona that reduces opsonization and extends circulation half-life. | mPEG-DSPE, mPEG-DMG, ALC-0159. [78] [9] |
| Polyvinyl Alcohol (PVA) | A stabilizer and surfactant used in the double emulsion method to prevent coalescence and control particle size. [79] | Varies by molecular weight and degree of hydrolysis. |
| Cationic Lipids | Essential for complexing with nucleic acids (DNA, mRNA) in lipid nanoparticles (LNPs), enabling efficient encapsulation and delivery. [9] | Ionizable lipids (e.g., in COVID-19 mRNA vaccines). |
| Ammonium Sulfate | Used to create a transmembrane pH gradient for the active (remote) loading of weakly basic drugs into liposomes, dramatically increasing encapsulation efficiency. [23] [80] | Used in buffer solutions for gradient formation. |
The comparative analysis of liposomal and polymeric nanoparticle formulations reveals a nuanced landscape where neither platform is universally superior. Instead, they offer complementary strengths, making them suitable for different therapeutic objectives. Liposomes stand out for their proven biocompatibility, clinical track record, and sophisticated active loading techniques for ionizable drugs. Their primary challenges lie in long-term stability and overcoming immune recognition. In contrast, polymeric nanoparticles excel in the tunability of their drug release profiles, the robustness of their matrix, and their potential for high encapsulation efficiencies, particularly for biologics and hydrophilic agents. Their translation, however, is more frequently hampered by manufacturing and reproducibility challenges related to polymer synthesis and processing.
The future of nanomedicine lies in leveraging these insights for rational design. For liposomes, research is focused on developing next-generation stealth coatings beyond PEG, optimizing targeting ligands, and improving storage stability. For polymeric nanoparticles, the priority is to develop scalable and reproducible synthesis methods, engineer polymers with predictable and safe degradation profiles, and design smart systems that actively respond to disease-specific stimuli. As the field matures, a shift from a purely particle-centric view to an integrated approach that considers the final formulation, manufacturing, and regulatory strategy from the earliest stages of development will be crucial to bridge the translational gap and deliver on the promise of nanomedicine for patients.
The evolution of drug delivery systems has been fundamentally transformed by the advent of nanocarrier technologies, with liposomal formulations and polymeric nanoparticles (PNPs) at the forefront. Liposomal formulations such as Doxil and AmBisome represent the first generation of clinically successful nanomedicines, leveraging passive targeting through the Enhanced Permeability and Retention (EPR) effect to improve drug bioavailability and reduce systemic toxicity [84] [8]. In parallel, polymeric nanoparticle (PNP) technologies have emerged as highly tunable alternatives, offering superior control over drug release kinetics, enhanced stability, and versatile functionalization for active targeting [38] [85]. This guide provides a comparative analysis of these platforms, focusing on their clinical performance, underlying experimental data, and patented technological advances that address persistent challenges in oncology and anti-infective therapy. The objective comparison herein is framed within the broader context of formulating more effective, safe, and targeted therapeutic interventions for researchers and drug development professionals.
Clinical Success Stories: Pioneering Liposomal Formulations
Doxil: A Paradigm Shift in Cancer Chemotherapy
Doxil (liposomal doxorubicin) stands as a landmark in cancer nanomedicine, specifically engineered to overcome the severe cardiotoxicity and dose-limiting side effects associated with conventional doxorubicin [84]. Its clinical success is underpinned by a unique drug loading mechanism and structural design.
- Formulation and Mechanism: Doxil employs a remote (active) loading method that utilizes an ammonium sulfate gradient across the liposomal membrane. This technique achieves exceptional drug-loading efficiency exceeding 90%, as doxorubicin diffuses into the liposome in its neutral form and becomes protonated and trapped as sulfate salts within the acidic core [84].
- Pharmacokinetics and Targeting: The formulation is stabilized by a polyethylene glycol (PEG) coating, creating a "stealth" characteristic that prolongs circulation time and facilitates passive accumulation in tumor tissues via the EPR effect [84] [8]. This results in a significantly improved therapeutic index by enhancing tumor-specific drug delivery while minimizing exposure to healthy tissues, particularly the heart.
Table 1: Key Formulation and Clinical Performance Characteristics of Doxil
| Attribute | Specification/Outcome | Clinical Impact |
|---|---|---|
| Loading Method | Remote loading via ammonium sulfate gradient [84] | High loading efficiency (>90%), stable encapsulation |
| Key Excipient | Polyethylene Glycol (PEG) [84] | "Stealth" property; prolonged circulation; reduced RES uptake |
| Targeting Mechanism | Passive targeting via EPR effect [84] | Enhanced drug accumulation in tumor tissues |
| Primary Clinical Benefit | Significant reduction in cardiotoxicity [84] | Enables higher cumulative doses; improved patient safety |
AmBisome and Fungisome: Liposomal Amphotericin B for Systemic Fungal Infections
Amphotericin B is a potent broad-spectrum antifungal agent, but its clinical use is limited by severe nephrotoxicity and infusion-related reactions. Liposomal encapsulation has been a successful strategy to mitigate these toxicities, with AmBisome and Fungisome serving as key comparative examples [86] [87].
A direct head-to-head comparative study in a murine model of cutaneous leishmaniasis revealed critical differences in their safety and efficacy profiles [86] [87]. Intravenous administration at 15 mg/kg showed clear signs of toxicity for Fungisome, while AmBisome was well-tolerated at the same dose. Furthermore, the anti-leishmanial efficacy of Fungisome was found to be inferior to AmBisome at equivalent doses (5 and 10 mg/kg), with higher ED50 values for both parasite load and lesion size. This was correlated with lower accumulation of the active drug within the infected lesions for Fungisome [86] [87].
Table 2: Comparative Efficacy and Toxicity of Liposomal Amphotericin B Formulations [86] [87]
| Parameter | AmBisome (A) | Fungisome (F) |
|---|---|---|
| Toxicity at 15 mg/kg | No clear signs of toxicity | Clear signs of toxicity observed |
| ED50 (Parasite Load) | 3.0 mg/kg | 4.0 mg/kg |
| ED50 (Lesion Size) | 8.8 mg/kg | 12.8 mg/kg |
| Drug Accumulation in Lesion | Higher levels | Lower levels |
| Inferred Therapeutic Index | Higher | Lower |
Advanced and Patented PNP Technologies
While liposomes have proven successful, polymeric nanoparticles offer distinct advantages, including versatile polymer chemistry for tailored drug release, convenient surface modification, and high stability [38] [85]. Recent patented technologies highlight the innovative approaches in this field.
"Click" Amphotericin B Prodrug Nanoformulations
A breakthrough in PNP technology for antifungal therapy involves a novel telodendrimer (TD) scaffold functionalized with multiple phenylboronic acid (PBA) moieties for "Click" prodrug conjugation of Amphotericin B (AmB) [88].
- Technology Overview: This platform utilizes reversible boronate ester chemistry to conjugate AmB directly to the telodendrimer without catalysts or organic solvents. The process is rapid, high-yielding, and avoids the need for dialysis purification, simplifying quality control for clinical translation [88].
- Stimuli-Responsive Release: The boronate linkage is stable in circulation but cleaves in response to acidic pH and elevated reactive oxygen species (ROS) levels common at sites of infection and inflammation, enabling targeted drug release [88].
- Experimental Outcomes: In vivo studies demonstrated that a single dose of the lead candidate, AmB-PEG5kBA4, was superior to both Fungizone and AmBisome in treating systemic fungal infections in immunocompromised mice. It also exhibited a maximum tolerated dose (MTD) comparable to AmBisome, representing a more than 20-fold increase over the conventional Fungizone formulation [88].
Polymeric Micelles for Doxorubicin Delivery
To address the limitations of Doxil, such as poor release control and instability, advanced polymeric micelles have been developed [84]. These systems focus on improving Drug Loading Capacity (DLC) and incorporating stimuli-responsive mechanisms.
- Enhanced Drug Loading: Conventional PEG-PLA or PEG-PCL micelles typically have a DLC of 5-10%. Recent strategies, such as co-micellization of polymers like MPEG-PDEAEMA and MPEG-PCL, have successfully increased DLC to 20-30% [84].
- Stimuli-Responsive Release: These smart PNPs are engineered to release their payload in response to specific tumor microenvironment triggers, such as low pH (pH-responsive) or high glutathione levels (redox-responsive) [84] [85]. This provides superior control over drug release compared to first-generation liposomes.
Diagram 1: Workflow of "Click" AmB-TD Prodrug Nanoformulation. The process involves catalyst-free conjugation via boronate chemistry, self-assembly into nanoparticles, and triggered drug release at the target site in response to acidic pH and reactive oxygen species (ROS) [88].
Comparative Analysis: Liposomal vs. Polymeric Nanoparticle Platforms
The choice between liposomal and polymeric nanocarriers involves a trade-off between the proven clinical track record of liposomes and the superior tunability and potential of advanced PNPs.
Table 3: Platform Comparison: Liposomal Formulations vs. Advanced Polymeric Nanoparticles
| Characteristic | Liposomal Formulations (Doxil, AmBisome) | Advanced Polymeric Nanoparticles (PNPs) |
|---|---|---|
| Targeting Mechanism | Primarily passive (EPR effect) [84] [8] | Passive (EPR) + Active (ligands) + Stimuli-responsive [84] [85] |
| Drug Release Control | Limited; relies on diffusion and liposome stability [84] | High; tunable via polymer chemistry and stimuli-responsive design [84] [85] |
| Drug Loading Capacity | High for specific drugs (e.g., via remote loading) [84] | Variable; typically lower, but advanced systems achieve 20-30% DLC [84] |
| Manufacturing & Scalability | Well-established, but complex for PEGylated liposomes [8] | Evolving; "Click" chemistry offers simpler, catalyst-free processes [88] |
| Clinical Translation | Extensive, multiple approved drugs [84] [87] | Emerging; several in clinical trials, facing heterogeneity and PK challenges [84] [81] |
The Scientist's Toolkit: Essential Research Reagents and Materials
The development and evaluation of nanocarrier systems rely on a specific set of reagents and analytical techniques. The following table details key materials used in the featured experiments and their functions.
Table 4: Essential Research Reagents and Materials for Nanocarrier Development
| Reagent / Material | Function / Role | Example from Featured Research |
|---|---|---|
| Polyethylene Glycol (PEG) | Provides "stealth" properties; reduces opsonization and extends circulation half-life [84] [8] | Component of Doxil and functionalized telodendrimers [84] [88] |
| Ammonium Sulfate | Creates a transmembrane gradient for active remote loading of drugs into liposomes [84] | Critical for high-efficiency loading of doxorubicin in Doxil [84] |
| Phenylboronic Acid (PBA) | Forms reversible boronate ester bonds with cis-diols; enables "Click" drug conjugation [88] | Key functional group on telodendrimer for conjugating Amphotericin B [88] |
| Telodendrimer (TD) | A linear-dendritic hybrid polymer serving as a versatile, well-defined nanoscaffold [88] | Core platform for attaching PBA and conjugating AmB in prodrug nanoformulations [88] |
| Poly(lactic-co-glycolic acid) (PLGA) | A biodegradable polymer used for controlled drug release in PNPs [89] | Used in microspheres and nanoparticles for sustained release applications [89] |
The direct comparison of Fungisome and AmBisome underscores that not all liposomal products are equivalent, with significant differences in safety and efficacy arising from formulation details [86] [87]. While Doxil and AmBisome remain foundational clinical success stories, the future of nanodrug delivery is being shaped by advanced PNP technologies. These include "Click" chemistry-based prodrugs and stimuli-responsive micelles that offer superior targeting, enhanced drug loading, and controlled release [84] [88]. For researchers, the ongoing challenge lies in optimizing these sophisticated PNP systems for manufacturing scalability, reproducible pharmacokinetics, and ultimately, successful clinical translation to provide new, more effective treatment paradigms.
In the evolving landscape of nanomedicine, polymeric nanoparticles (PNPs) and liposomes represent two of the most prominent platforms for targeted drug delivery. While both systems aim to enhance therapeutic efficacy and reduce side effects, their fundamental properties, capabilities, and clinical translation potential differ significantly. The choice between these nanocarriers is not trivial but must be guided by a systematic assessment of the active pharmaceutical ingredient's (API) characteristics, the target indication's biological constraints, and practical development considerations. This framework synthesizes current evidence and experimental methodologies to provide researchers with a structured approach for selecting the optimal nanocarrier system based on specific project requirements. The high failure rates of nanomedicine candidates—with only an estimated 50-80 products achieving global approval by 2025 despite thousands of preclinical candidates—highlight the critical importance of early-stage platform selection [78]. By aligning API properties and therapeutic goals with the inherent strengths of each delivery system, development teams can increase their probability of technical success while avoiding costly late-stage failures.
Comparative Analysis: Fundamental Properties of PNPs and Liposomes
Table 1: Core characteristics of polymeric nanoparticles and liposomes
| Parameter | Polymeric Nanoparticles (PNPs) | Liposomes |
|---|---|---|
| Structure | Solid polymer matrix (e.g., PLGA, chitosan) | Phospholipid bilayers enclosing aqueous core |
| Size Range | 10-1000 nm [1] | 50-200 nm (ideal for drug delivery) [90] |
| Drug Loading Capacity | High for hydrophobic drugs; tunable via polymer selection [22] [24] | Hydrophilic drugs (aqueous core); hydrophobic drugs (lipid bilayer) [23] [90] |
| Encapsulation Efficiency | Varies by method and polymer; generally high for compatible drugs | Can be optimized via active loading (e.g., ammonium sulfate gradient) [23] |
| Release Kinetics | Controlled degradation rate; sustained release profiles [22] [78] | Often biphasic (initial burst then sustained); can be stimuli-responsive [91] [23] |
| Surface Modification | Versatile chemistry for PEGylation, targeting ligands [22] [1] | PEGylation common; ligand conjugation to lipid heads [90] [8] |
| Scalability & Manufacturing | Batch-to-batch variability challenges; complex GMP scaling [78] | Established production methods (thin-film hydration, REV) [90] |
| Regulatory Precedent | Limited approved products [78] | ~15 FDA-approved products [90] |
| Key Advantages | Superior stability; controlled release profiles; material versatility [1] [24] | Clinical validation; biocompatibility; efficient drug loading mechanisms [23] [92] |
Table 2: API compatibility and formulation considerations
| API Characteristic | Recommended System | Rationale & Experimental Evidence |
|---|---|---|
| Hydrophobic Drugs | PNPs (especially polymeric micelles) | Polymer matrix provides hydrophobic domains for encapsulation; demonstrated with paclitaxel formulations [78] |
| Hydrophilic Drugs | Liposomes | Aqueous core provides natural compartment; high loading via gradient methods [23] [90] |
| Amphiphilic Drugs | Liposomes | Can partition between bilayer and aqueous core based on properties [90] |
| Nucleic Acids (DNA, RNA) | Both (LNPs for RNA; cationic polymers for DNA) | LNPs clinically validated for mRNA; cationic PNPs for DNA binding [78] [1] |
| Proteins/Peptides | PNPs (especially biodegradable polymers) | Protection from degradation; controlled release maintains stability [1] [24] |
| Ionizable Weak Bases/Acids | Liposomes | Excellent loading via pH/ion gradients (e.g., ammonium sulfate) [23] |
Decision Framework: Matching Platform to Application
The selection between PNPs and liposomes requires evaluating multiple intersecting factors related to the API, target disease, and development constraints. The following diagram illustrates the key decision pathways:
Diagram 1: Decision pathway for selecting between PNPs and liposomes
Key Decision Factors
API Physicochemical Properties
The chemical nature of the therapeutic agent profoundly influences carrier selection. Liposomes demonstrate superior capabilities for hydrophilic compounds, which can be efficiently loaded into their aqueous core using established gradient methods [23]. For example, the ammonium sulfate gradient technique enables high loading of weakly basic drugs through active accumulation mechanisms. Conversely, PNPs excel with hydrophobic APIs that can be effectively incorporated into the polymer matrix, with materials like PLGA providing sustained release profiles that maintain therapeutic levels over extended periods [78] [24]. For nucleic acid delivery, lipid nanoparticles (LNPs) have established clinical success with mRNA vaccines, while cationic polymers remain valuable for DNA delivery applications [78] [1].
Release Kinetics Requirements
The desired drug release profile represents another critical consideration. PNPs offer superior control over extended release durations, from days to weeks, through careful polymer selection and fabrication tuning. The degradation rate of biodegradable polymers like PLGA directly governs release kinetics, enabling precise temporal control [78]. Liposomes typically exhibit biphasic release patterns and excel in stimuli-responsive applications where immediate, site-specific release is desired. Advanced liposomal designs demonstrate triggered release in response to ultrasound [91], pH changes [23], or enzymatic activity [8], making them ideal for targeted burst delivery in response to specific biological triggers.
Target Indication and Biological Barriers
The pathological environment and associated biological barriers significantly impact platform selection. Liposomes leverage the enhanced permeability and retention (EPR) effect for passive tumor targeting, although this effect shows considerable heterogeneity in human patients [78] [90]. Their clinical validation in oncology is substantial, with numerous approved products. PNPs show particular promise for crossing challenging barriers like the blood-brain barrier (BBB), where their surface functionality and controlled release characteristics can enhance CNS delivery [1]. For inflammatory conditions, both systems can be effective, though liposomes may benefit from inherent accumulation at inflamed sites due to vascular permeability similarities to the EPR effect [8].
Experimental Protocols and Characterization Methods
Key Characterization Techniques
Table 3: Essential characterization methods for nanocarrier development
| Characterization Method | Parameters Measured | Protocol Highlights | Relevance to Platform Selection |
|---|---|---|---|
| Dynamic Light Scattering (DLS) | Size, polydispersity index (PDI) | Dilute in appropriate buffer; measure at 25°C; multiple measurements | Critical for both systems; size affects biodistribution and EPR [1] [23] |
| Nuclear Magnetic Resonance (NMR) Spectroscopy | Polymer conversion, drug conjugation efficiency | Use deuterated solvents; track monomer disappearance | Particularly valuable for PNPs to confirm polymer structure and drug loading [1] |
| Transmission Electron Microscopy (TEM) | Morphology, lamellarity, structural integrity | Negative staining with uranyl acetate; cryo-EM for native state | Essential for confirming liposomal structure and PNP morphology [91] [1] |
| HPLC for Encapsulation Efficiency | Drug loading, encapsulation efficiency | Separate free drug; disrupt particles for total drug | Standard method for both systems; critical for formulation optimization [23] |
| In Vitro Release Studies | Release kinetics, mechanism | Dialysis method; sink conditions; physiological pH/temperature | Key for comparing sustained release capabilities [22] [24] |
Protocol: Ultrasound-Responsive Liposomal Drug Release Assessment
Recent advances in stimuli-responsive liposomes demonstrate the importance of specialized release testing protocols. The following methodology is adapted from acoustically activatable liposomes (AALs) that incorporate generally regarded as safe (GRAS) excipients to enhance ultrasound responsiveness [91]:
Materials and Equipment:
- Liposomes prepared with 5% added sucrose in internal buffer
- Therapeutic ultrasound system (e.g., clinical FUS system)
- Temperature-controlled incubation chamber (37°C)
- HPLC system with appropriate detection
- Dialysis membrane or centrifugal filters for separation
Procedure:
- Prepare liposomal formulation with internal sucrose gradient (5% w/v) to alter acoustic properties of the core medium
- Incubate liposomes in plasma at 37°C to simulate physiological conditions
- Apply pulsed, low-intensity ultrasound (mechanical index = 1.8, in situ peak negative pressure = 0.9 MPa, frequency = 250 kHz)
- Maintain temperature controls to distinguish mechanical from thermal release mechanisms
- Separate released drug from encapsulated drug using centrifugal filtration (30kDa MWCO)
- Quantify drug content in release medium using validated HPLC methods
- Compare ultrasound-treated samples to non-sonicated controls incubated under identical conditions
Validation Metrics:
- Significant drug release (approximately 40%) with sucrose incorporation versus minimal release without sucrose
- No signs of inertial cavitation (confirmed by absence of ultraharmonics in acoustic monitoring)
- Minimal heating (<0.2°C) to confirm mechanical rather than thermal mechanism
- Stability in plasma without ultrasound (less than 10% premature release over 1 hour)
This protocol demonstrates how specialized release testing can validate stimulus-responsive behavior, a key consideration for certain target indications.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key reagents and materials for nanocarrier development
| Reagent/Material | Function | Application Examples |
|---|---|---|
| DSPC (Lipid) | Primary phospholipid for bilayer formation | Liposome structural component [90] |
| Cholesterol | Membrane stabilization, rigidity | Liposome formulation (30-50% ratio) [90] |
| PLGA Polymer | Biodegradable polymer matrix | PNPs for sustained release [78] [24] |
| DSPE-PEG(2000) | Stealth properties, circulation extension | PEGylation of both PNPs and liposomes [78] [8] |
| Ammonium Sulfate | Active loading gradient for weak bases | Liposomal loading of doxorubicin, cisplatin [23] |
| Sucrose | Acoustic property modification, cryoprotection | Ultrasound-responsive liposomes [91] |
| Chitosan | Mucoadhesive properties, positive charge | PNPs for mucosal delivery [78] [24] |
Clinical Translation and Regulatory Considerations
The translational pathway differs substantially between PNPs and liposomes, impacting development timelines and resource allocation. Liposomes benefit from a well-established regulatory precedent with approximately 15 FDA-approved products and clearer Chemistry, Manufacturing, and Controls (CMC) requirements [92] [90]. Their production methods, such as thin-film hydration and reverse-phase evaporation, are more standardized and scalable. However, challenges remain regarding sterilization, stability during storage, and potential immune reactions to PEGylated components [23].
PNPs face greater translational hurdles despite their design flexibility. Batch-to-batch variability presents significant manufacturing challenges, and the correlation between in vitro characterization and in vivo performance remains less predictable [78]. The regulatory pathway for novel polymeric materials is more complex, requiring extensive safety and biodegradation profiling. However, PNPs offer advantages in storage stability and controlled release profiles that can justify their development for specific applications where liposomes are suboptimal.
The recent failure of advanced candidates like BIND-014 (targeted docetaxel nanoparticles) despite promising preclinical data highlights the translational gap particularly affecting complex targeted systems [78]. This underscores the importance of early attention to scalability and manufacturability in platform selection.
The selection between polymeric nanoparticles and liposomes represents a foundational decision in nanomedicine development that should be guided by systematic assessment of API properties, target indication requirements, and practical development constraints. Liposomes offer clinical validation, efficient loading mechanisms for specific drug classes, and well-characterized manufacturing processes. PNPs provide superior control over release kinetics, enhanced stability, and material versatility for challenging APIs.
Future developments will likely blur the distinctions between these platforms through hybrid approaches that combine advantageous properties of both systems. Emerging trends include stimuli-responsive designs that activate drug release at target sites [91], non-PEG stealth alternatives to address immunogenicity concerns [78] [8], and personalized approaches based on patient-specific transport biomarkers. The increasing integration of artificial intelligence in nanoparticle optimization and characterization will further refine selection algorithms and design parameters [22] [38].
By applying this structured framework during early development stages, researchers can make informed platform selections that align with their therapeutic objectives while anticipating downstream development challenges, ultimately increasing the probability of successful clinical translation.
Conclusion
Both polymeric nanoparticles and liposomal formulations present powerful, yet distinct, platforms for advanced drug delivery. PNPs often offer superior structural stability and tunable drug release profiles, while liposomes excel in biocompatibility and efficient encapsulation of diverse payloads. The choice between them is not a matter of superiority but of strategic alignment with the specific therapeutic agent, target disease, and delivery route. Future advancements are poised to blur the lines between these technologies, with a clear trend toward intelligent, stimuli-responsive, and hybrid nanocarriers. The integration of artificial intelligence in design optimization, alongside a stronger focus on scalable and reproducible manufacturing processes, will be pivotal in translating more nanomedicines from the laboratory to the clinic, ultimately enabling more effective and personalized therapies.
References
- 1. Characterisation of polymeric nanoparticles for drug delivery [https://pubs.rsc.org/en/content/articlehtml/2025/nr/d5nr00071h]
- 2. Biodegradable Polymeric Nanoparticle-Based Drug ... [https://www.mdpi.com/2073-4360/16/17/2536]
- 3. Chitosan nanoparticles: Green synthesis, biological ... [https://www.sciencedirect.com/science/article/pii/S2590006425009299]
- 4. A dataset on formulation parameters and characteristics of ... [https://www.nature.com/articles/s41597-025-04621-9]
- 5. PEGylated PLGA nanoparticles: unlocking advanced ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02410-x]
- 6. Green nanocarriers and Biodegradable Systems for ... [https://www.sciencedirect.com/science/article/abs/pii/S1773224725006112]
- 7. More than a delivery system: the evolving role of lipid-based ... [https://pubs.rsc.org/en/content/articlehtml/2025/nr/d4nr04508d]
- 8. Advances in Liposomal Drug Delivery - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC12298344/]
- 9. Liposomes vs. Lipid Nanoparticles: Which Is Best for Drug ... [https://www.biochempeg.com/article/122.html]
- 10. A Review on Polymer and Lipid-Based Nanocarriers and Its ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8671830/]
- 11. Polymeric Nanoparticle Versus Liposome Formulations [https://pubmed.ncbi.nlm.nih.gov/33156405/]
- 12. A comparison study of lipid and polymeric nanoparticles in ... [https://www.sciencedirect.com/science/article/pii/S0378517321005299]
- 13. Cholesterol Within Phosphatidylcholine Liposomes Attenuates ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12238836/]
- 14. Unveiling the impact of membrane fluidity in shaping lipid- ... [https://www.sciencedirect.com/science/article/abs/pii/S0005273625000550]
- 15. The gold nanoparticle–lipid membrane synergy for ... [https://pubs.rsc.org/en/content/articlehtml/2025/nh/d5nh00292c]
- 16. A machine learning workflow to accelerate the design of in ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00112a]
- 17. Comparison between multilamellar and unilamellar ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1454112/]
- 18. Evaluation and Comparison of Solid Lipid Nanoparticles ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8063941/]
- 19. Polymeric Lipid Hybrid Nanoparticles (PLNs) as Emerging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8399620/]
- 20. Molecularly imprinted polymer-based core-shells (solid vs ... [https://www.sciencedirect.com/science/article/abs/pii/S0925400516320809]
- 21. Comprehensive analysis of lipid nanoparticle formulation ... [https://www.sciencedirect.com/science/article/pii/S2590156724000550]
- 22. Polymeric Nanoparticles in Targeted Drug Delivery: Unveiling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11991310/]
- 23. Liposomal Formulations: A Recent Update - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11769406/]
- 24. Current updated review on preparation of polymeric ... [https://www.sciencedirect.com/science/article/pii/S294982952300013X]
- 25. Polymeric Nanoparticles for Delivery of Natural Bioactive ... [https://www.mdpi.com/2073-4360/15/5/1123]
- 26. Strategic advances in liposomes technology: translational ... [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-025-03660-z]
- 27. pH-responsive high stability polymeric nanoparticles for ... [https://www.nature.com/articles/s42003-020-0817-4]
- 28. Development and characterization of liposomal ... [https://www.nature.com/articles/s41598-024-57663-1]
- 29. Microfluidic formulation of nanoparticles for biomedical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8752123/]
- 30. Microfluidics for PLGA nanoparticle synthesis: a review [https://elveflow.com/microfluidic-reviews/microfluidics-for-plga-nanoparticle-synthesis-a-review/]
- 31. Nanomanufacturing through microfluidic-assisted ... [https://www.sciencedirect.com/science/article/abs/pii/S0378517317309560]
- 32. Recent advances in nanoprecipitation - RSC Publishing [https://pubs.rsc.org/en/content/articlehtml/2025/sm/d5sm00006h]
- 33. A Comprehensive Review on Novel Liposomal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8790012/]
- 34. A Review of Liposomes as a Drug Delivery System [https://pmc.ncbi.nlm.nih.gov/articles/PMC8879473/]
- 35. Methods of Liposomes Preparation: Formation and Control ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8955843/]
- 36. Basic Methods for Preparation of Liposomes and Studying ... [https://www.mdpi.com/1422-0067/22/12/6547]
- 37. Conventional Liposomal Formulation Methods [https://encyclopedia.pub/entry/44196]
- 38. Advances in nanoparticles in targeted drug delivery–A review [https://www.sciencedirect.com/science/article/pii/S2666845925001163]
- 39. Review Advantages and limitations of nanostructures for ... [https://advances.umw.edu.pl/en/article/2025/34/3/447/]
- 40. Advances in liposomal nanotechnology: from concept to clinics [https://pubs.rsc.org/en/content/articlehtml/2024/pm/d4pm00176a]
- 41. PEGylation in Pharmaceutical Development - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC11390148/]
- 42. The benefits and risks of PEGylation in nanomedicine [https://www.nature.com/articles/s41565-025-01951-y]
- 43. Questioning the Use of PEGylation for Drug Delivery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4051498/]
- 44. Stimuli-Responsive Nanomedicines for the Treatment of Non ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12045021/]
- 45. Conventional vs PEGylated loaded liposomal formulations ... [https://www.sciencedirect.com/science/article/pii/S0378517324003119]
- 46. Polymeric Nanoparticles for Targeted Lung Cancer Treatment [https://pmc.ncbi.nlm.nih.gov/articles/PMC12472848/]
- 47. Next-Generation Liposomal Drug Delivery Systems [https://www.preprints.org/manuscript/202501.1875]
- 48. Strategies for delivering drugs across the blood-brain ... [https://www.frontiersin.org/journals/drug-delivery/articles/10.3389/fddev.2025.1644633/full]
- 49. Engineered Liposomal Delivery of Human ACE2 Across the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12566711/]
- 50. Crossing the blood–brain barrier: advances in dendrimer ... [https://pubs.rsc.org/en/content/articlelanding/2025/nr/d5nr02548f]
- 51. Polymeric nanoparticles in the treatment of dermatological ... [https://www.sciencedirect.com/science/article/abs/pii/B9780443363269000198]
- 52. Topical and transdermal lipid-polymer hybrid nanoparticles ... [https://pubmed.ncbi.nlm.nih.gov/40802031/]
- 53. Insights into Liposomal and Gel-Based Formulations for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12027463/]
- 54. Liposomal Formulations: A Recent Update [https://pubmed.ncbi.nlm.nih.gov/39861685/]
- 55. Colloidal stability of polymeric nanoparticles in biological ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3496558/]
- 56. Comparison of liposomal drug formulations for transdermal ... [https://www.sciencedirect.com/science/article/pii/S092809871730369X]
- 57. Effect of polymeric stabilizers on the size and ... [https://www.sciencedirect.com/science/article/pii/S1319016423001925]
- 58. Toward the Scalable, Rapid, Reproducible, and Cost ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11211002/]
- 59. Factors affecting drug encapsulation and stability of lipid– ... [https://www.sciencedirect.com/science/article/abs/pii/S0927776511001111]
- 60. The Ultimate Chemical Manufacturing Scale-Up Checklist [https://optimation.rebuildmanufacturing.com/blogs/the-ultimate-chemical-manufacturing-scale-up-checklist/]
- 61. An overview of liposomal nano-encapsulation techniques ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7447741/]
- 62. Overcoming Biosimilar Scaling Challenges [https://www.pharmtech.com/view/overcoming-biosimilar-scaling-challenges]
- 63. Nanoparticle Contract Manufacturing Market Forecast 2031 [https://www.transparencymarketresearch.com/nanoparticle-contract-manufacturing-market.html]
- 64. Emerging strategies against accelerated blood clearance ... [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-025-03209-0]
- 65. Rediscovery of mononuclear phagocyte system blockade ... [https://www.nature.com/articles/s41467-024-48838-5]
- 66. Review Biomimetic approaches against accelerated blood ... [https://www.sciencedirect.com/science/article/abs/pii/S0378517325005903]
- 67. Effects of complement inhibition on the ABC phenomenon ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7032085/]
- 68. Accelerated blood clearance phenomenon upon cross ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4437610/]
- 69. Polymeric micelles versus liposomes [https://atomfair.com/nanomaterial-primer/article.php?id=G49-880]
- 70. A comparative study of the merits of polymersomes in terms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6956745/]
- 71. Effects of nanoparticle size, shape, and zeta potential on ... [https://www.sciencedirect.com/science/article/abs/pii/S0378517324010330]
- 72. Quality by Design-Driven Zeta Potential Optimisation Study of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9503861/]
- 73. Practical Guidelines for the Characterization and Quality ... [https://pubmed.ncbi.nlm.nih.gov/29920294/]
- 74. Polymeric Nanoparticle Versus Liposome Formulations: Comparative Physicochemical and Metabolomic Studies as l-Carnitine Delivery Systems | AAPS PharmSciTech [https://link.springer.com/article/10.1208/s12249-020-01852-4]
- 75. Predicting PLGA nanoparticle size and zeta potential in ... [https://www.nature.com/articles/s41598-025-06872-3]
- 76. Tuning Liposome Stability in Biological Environments and ... [https://www.mdpi.com/2218-273X/13/1/59]
- 77. Nanomaterials in Drug Delivery: Leveraging Artificial ... [https://www.mdpi.com/1422-0067/26/22/11121]
- 78. Bridging the Gap: The Role of Advanced Formulation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12579833/]
- 79. Nanotechnology Meets superbugs [https://pmc.ncbi.nlm.nih.gov/articles/PMC12569084/]
- 80. Recent advances in liposome formulations for breast ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11071878/]
- 81. Advanced rational design of polymeric nanoparticles for ... [https://www.sciencedirect.com/science/article/abs/pii/S0168365925005607]
- 82. Smart Polymeric Nanoparticles for Targeted Delivery and ... [https://www.sciencedirect.com/science/article/pii/S2590183425000341]
- 83. Recent advances in polymer-based nanoparticles [https://pubmed.ncbi.nlm.nih.gov/41175039/]
- 84. Advances in Doxorubicin Chemotherapy: Emerging Polymeric ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12293504/]
- 85. Polymeric Nanoparticles in Targeted Drug Delivery [https://www.mdpi.com/2073-4360/17/7/833]
- 86. Comparative efficacy, toxicity and biodistribution of the ... [https://www.sciencedirect.com/science/article/pii/S2211320718300101]
- 87. Comparative efficacy, toxicity and biodistribution of the ... [https://pubmed.ncbi.nlm.nih.gov/29673889/]
- 88. “Click” Amphotericin B in Prodrug Nanoformulations for ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11923797/]
- 89. An Overview of Polymeric Nanoparticles-Based Drug Delivery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9893377/]
- 90. Targeted Liposomal Drug Delivery: Overview of the Current ... [https://www.mdpi.com/2075-1729/14/6/672]
- 91. Acoustically activatable liposomes as... | Nature Nanotechnology [https://www.nature.com/articles/s41565-025-01990-5?error=cookies_not_supported&code=691e76bd-0c58-4c47-8dd3-218b6e3fbab6]
- 92. A Review of Liposomes as a Drug Delivery System: Current Status of... [https://pubmed.ncbi.nlm.nih.gov/35209162/]