Viral vs. Non-Viral Gene Delivery: Principles, Applications, and Choosing the Right Vector for Therapeutics
This comprehensive guide examines the core principles of viral and non-viral gene delivery vectors, essential tools for researchers and drug developers in advanced therapeutics.
Viral vs. Non-Viral Gene Delivery: Principles, Applications, and Choosing the Right Vector for Therapeutics
Abstract
This comprehensive guide examines the core principles of viral and non-viral gene delivery vectors, essential tools for researchers and drug developers in advanced therapeutics. It provides a foundational understanding of vector biology, details practical methodologies for vector design and application, addresses common challenges with optimization strategies, and offers a comparative analysis for informed vector selection. The article synthesizes current trends to empower professionals in navigating the complex landscape of gene therapy and genetic medicine development.
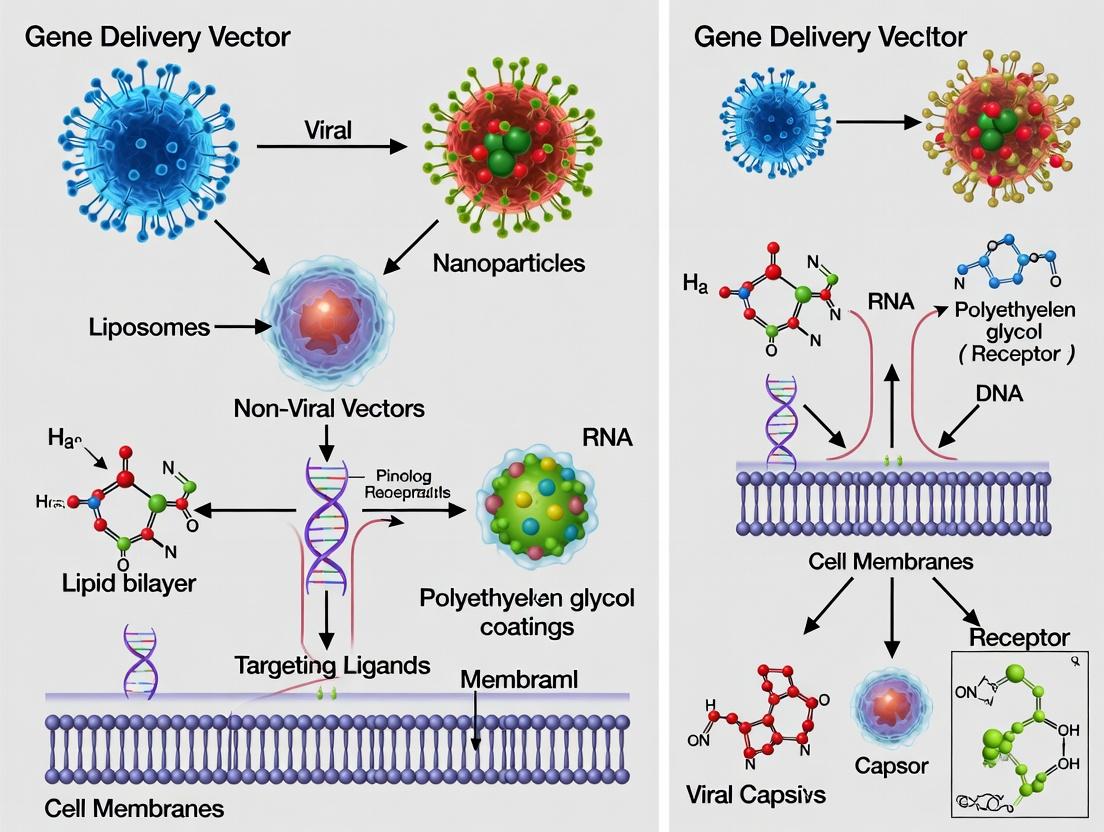
Gene Delivery Vectors 101: Core Principles and Biological Mechanisms Explained
Gene delivery is the process of introducing exogenous nucleic acids (DNA or RNA) into host cells to alter gene expression for research or therapeutic purposes. The ultimate goal is to achieve sufficient transfection or transduction efficiency to produce a desired phenotypic effect, such as correcting a genetic defect, inducing an immune response, or studying gene function. The fundamental challenge is overcoming cellular and systemic barriers—including enzymatic degradation, immune clearance, and the plasma membrane—to deliver genetic cargo safely and efficiently to target cells and tissues. This necessitates the use of specialized delivery vehicles known as vectors.
Core Barriers to Naked Nucleic Acid Delivery
Naked nucleic acids are rapidly degraded by serum nucleases and elicit immune responses. Their anionic charge and hydrophilic nature prevent passive diffusion across the hydrophobic lipid bilayer of the cell membrane. Even if internalized, they must escape endosomal degradation and, for DNA, reach the nucleus. Vectors are engineered systems designed to circumvent these barriers.
Vector Classification and Quantitative Comparison
Vectors are broadly categorized as viral or non-viral. The table below summarizes key quantitative metrics.
Table 1: Comparison of Major Gene Delivery Vector Systems
| Vector Type | Max. Cargo Capacity (kb) | Typical Transfection/Transduction Efficiency In Vitro | Immunogenicity | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Adenovirus | 7-8 (first gen) ~36 (HDAd) | High (>90% in permissive cells) | High | High titer, broad tropism, high efficiency | Strong immune response, transient expression |
| Adeno-Associated Virus (AAV) | ~4.7 | Moderate to High (cell-type dependent) | Low | Long-term expression, excellent safety profile, serotype tropism | Small cargo limit, potential pre-existing immunity |
| Lentivirus | ~8-10 | High (>80% in dividing & non-dividing) | Moderate | Genomic integration, long-term expression, broad tropism | Insertional mutagenesis risk, complex production |
| Gamma-Retrovirus | ~8 | High (in dividing cells only) | Moderate | Stable integration | Only transduces dividing cells, insertional risk |
| Lipid Nanoparticles (LNPs) | >10 | Moderate to High (varies with cell type) | Low to Moderate (reactogenic) | Scalable, versatile cargo (DNA, mRNA, siRNA), no viral risks | Potential cytotoxicity, transient expression, liver tropism in vivo |
| Polymeric Vectors (e.g., PEI) | >10 | Moderate | High (cytotoxicity) | Easy synthesis, high cargo capacity | High cytotoxicity, aggregation in serum |
| Physical Methods (Electroporation) | N/A (size-limited by pores) | High (but cell-type sensitive) | N/A | Direct delivery, no chemical carrier | High cell mortality, not suitable for all tissues in vivo |
Detailed Experimental Protocols for Vector Evaluation
Protocol:In VitroTransduction Efficiency Assay for Lentiviral Vectors
Objective: Quantify the percentage of cells successfully transduced by a GFP-encoding lentivirus.
Materials:
- HEK293T cells (or target cell line of interest)
- Complete growth medium
- Lentiviral supernatant (e.g., VSV-G pseudotyped, encoding GFP)
- Polybrene (hexadimethrine bromide, 8 mg/mL stock)
- Phosphate Buffered Saline (PBS)
- Flow cytometry buffer (PBS + 2% FBS)
- 4% Paraformaldehyde (PFA) in PBS (optional for fixation)
- 6-well tissue culture plates
- Flow cytometer
Procedure:
- Seed Cells: Plate 2 x 10^5 cells per well in a 6-well plate in 2 mL complete medium. Incubate at 37°C, 5% CO2 for 18-24 hours to reach ~70% confluency.
- Prepare Transduction Mix: In a sterile tube, dilute the lentiviral supernatant to the desired multiplicity of infection (MOI, e.g., 5, 10, 20) in 1 mL of fresh, serum-containing medium. Add Polybrene to a final concentration of 8 µg/mL to enhance viral attachment.
- Transduce Cells: Aspirate medium from the wells. Add the 1 mL transduction mix to each well. Include a negative control well with medium + Polybrene only.
- Incubate: Return plate to incubator for 6-8 hours.
- Refresh Medium: Carefully aspirate the transduction mix and replace with 2 mL fresh complete medium.
- Incubate and Express: Culture cells for 48-72 hours to allow for GFP expression.
- Harvest and Analyze: a. Wash cells with PBS. b. Detach cells using trypsin-EDTA or a gentle cell dissociation reagent. c. Neutralize with complete medium, transfer to flow cytometry tubes. d. Pellet cells at 300 x g for 5 min, wash with flow cytometry buffer, and resuspend in 300 µL buffer. (Optional: Fix cells with 4% PFA for 15 min on ice, then wash twice).
- Flow Cytometry: Analyze 10,000 events per sample on a flow cytometer using a 488 nm laser and a 530/30 nm bandpass filter. Gate on live, single cells. The percentage of GFP-positive cells in the transduced sample (minus background from the control) is the transduction efficiency.
Protocol: Formulation andIn VitroScreening of Lipid Nanoparticles (LNPs) for mRNA Delivery
Objective: Formulate and test LNPs encapsulating mRNA and assess delivery efficiency.
Materials:
- Ionizable lipid (e.g., DLin-MC3-DMA)
- Helper lipids: DSPC, Cholesterol, PEG-lipid (e.g., DMG-PEG2000)
- Ethanol (100%)
- mRNA of interest in 10 mM citrate buffer (pH 4.0)
- Microfluidic mixer (e.g., NanoAssemblr) or T-tube apparatus
- Dialysis cassettes (MWCO 10kDa)
- Phosphate Buffered Saline (PBS)
- HEK293 or HeLa cells
- Luciferase assay kit (if mRNA encodes luciferase)
- Ribogreen assay kit
Procedure:
- Prepare Lipid Stock: Dissolve ionizable lipid, DSPC, Cholesterol, and PEG-lipid at a molar ratio (e.g., 50:10:38.5:1.5) in ethanol to a total lipid concentration of 12.5 mM.
- Prepare Aqueous Phase: Dilute mRNA in citrate buffer to a concentration of 0.2 mg/mL.
- Formulation: a. For microfluidic mixing: Set the flow rate ratio of aqueous phase to ethanol phase to 3:1 (e.g., 12 mL/min aqueous, 4 mL/min lipid). Pump both solutions into the mixing chamber. The resulting LNP suspension is collected in a vial. b. For rapid mixing: Rapidly inject the ethanol lipid solution into the stirred aqueous mRNA solution using a pipette or syringe.
- Dialysis: Immediately transfer the crude LNP suspension to a dialysis cassette. Dialyze against 1 L PBS at 4°C for 18-24 hours to remove ethanol and exchange the buffer.
- Characterization: Measure particle size and polydispersity index (PDI) via dynamic light scattering, and zeta potential via electrophoretic light scattering.
- Encapsulation Efficiency: Use a Ribogreen assay. Measure total mRNA by lysing LNPs with 1% Triton X-100. Measure free/unencapsulated mRNA without lysis. Calculate encapsulation efficiency:
EE% = (1 - (Free mRNA/Total mRNA)) * 100. - In Vitro Transfection: a. Seed cells in a 96-well plate (1x10^4 cells/well). b. After 24h, treat cells with LNP-mRNA formulations at various doses (e.g., 10-200 ng mRNA/well). c. Incubate for 24-48 hours. d. Assay for function: If mRNA encodes luciferase, lyse cells and measure luminescence. Normalize to total protein (BCA assay) or cell number.
Visualizations
Pathways of Cellular Uptake and Intracellular Trafficking for Vectors
Diagram Title: Vector Uptake and Intracellular Trafficking Pathways
Workflow for Developing and Testing a Gene Delivery Vector
Diagram Title: Gene Delivery Vector R&D Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for Gene Delivery Research
| Reagent/Material | Primary Function | Example Product/Component |
|---|---|---|
| Packaging Plasmids (Viral) | Provide essential viral proteins in trans for producing replication-incompetent viral vectors. | psPAX2 (lentiviral gag/pol/rev), pMD2.G (VSV-G envelope), pAdVAntage (adenoviral helper). |
| Transfection Reagent | Facilitate in vitro delivery of plasmid DNA or RNA during vector production or screening. | Polyethylenimine (PEI), Lipofectamine 3000, FuGENE HD. |
| Polybrene | A cationic polymer that reduces charge repulsion, enhancing viral vector attachment to target cells. | Hexadimethrine bromide, typically used at 4-8 µg/mL. |
| Ionizable Cationic Lipid | Key component of LNPs; positively charged at low pH to complex nucleic acids and promote endosomal escape. | DLin-MC3-DMA, SM-102, ALC-0315. |
| PEGylated Lipid | Stabilizes LNP formulations, reduces aggregation, modulates pharmacokinetics in vivo. | DMG-PEG2000, DSPE-PEG2000. |
| Size Exclusion Chromatography Resin | Purifies viral vectors or LNPs by separating them from free proteins, nucleic acids, or aggregates. | Sepharose 4FF, Superose 6 Increase columns. |
| qPCR/PCR Reagents for Titering | Quantifies viral genomic titer (vector genomes/mL) for dose standardization. | SYBR Green or TaqMan assays targeting viral backbone (e.g., WPRE in lentivirus). |
| Luciferase Reporter Plasmid/mRNA | Standardized cargo to quantify delivery efficiency via bioluminescence signal. | pGL4 luciferase vectors, CleanCap FLuc mRNA. |
| Cell Line with Stable Reporter | Stably expresses a fluorescent or luminescent protein upon successful delivery/editing. | HEK293-GFP, HeLa-Luc2, or CRE-responsive tdTomato reporter cells. |
| In Vivo Imaging System | Enables non-invasive, longitudinal tracking of gene expression in live animals. | IVIS Spectrum (PerkinElmer) or equivalent for bioluminescence/fluorescence. |
Within the broader thesis on basic principles of gene delivery vectors, viral vectors represent the most mature and clinically proven platform for gene transfer. Their fundamental biology dictates their experimental and therapeutic applications. This guide details the core characteristics, mechanisms, and methodologies for the four primary viral vector toolkits.
Fundamental Biology & Quantitative Comparison
Viral vectors are engineered from wild-type viruses by deleting pathogenic genetic elements and preserving the components necessary for gene delivery. The choice of vector is dictated by payload capacity, tropism, duration of expression, and immunogenicity.
Table 1: Core Characteristics of Major Viral Vectors
| Property | Adenovirus (AdV) | Adeno-Associated Virus (AAV) | Lentivirus (LV) | Retrovirus (γ-Retrovirus, RV) |
|---|---|---|---|---|
| Genome Type | dsDNA | ssDNA | ssRNA (+) | ssRNA (+) |
| Packaging Capacity | ~8-36 kb (gutted) | ~4.7 kb | ~8-10 kb | ~8-10 kb |
| Integration | Episomal | Predominantly episomal (low-freq. site-specific) | Integration (random) | Integration (random) |
| In Vivo Immune Response | High (capsid & transgene) | Generally low (capsid-specific) | Low to moderate | Low to moderate |
| Titers (Common Range) | 10^10 - 10^13 VP/mL | 10^11 - 10^13 VG/mL | 10^7 - 10^9 TU/mL | 10^6 - 10^8 TU/mL |
| Transduction Efficiency (In Vitro) | High for many dividing/non-dividing cells | Variable; high with optimized serotype | High for dividing & non-dividing cells | High for dividing cells only |
| Onset of Expression | Rapid (hours-days) | Slow (days-weeks) | Moderate (days) | Moderate (days) |
| Expression Duration | Transient (weeks) | Long-term (years in post-mitotic tissue) | Long-term (stable integration) | Long-term (stable integration) |
| Key Clinical Application | Vaccines, oncolytic therapy | In vivo gene therapy (e.g., Luxturna, Zolgensma) | Ex vivo cell therapy (e.g., CAR-T), HSC gene therapy | Ex vivo HSC gene therapy (historical) |
Key Experimental Protocols
Protocol: Production of Recombinant AAV via Triple Transfection in HEK293 Cells
This is the most common method for research-grade AAV production.
- Cell Seeding: Seed HEK293 cells (which supply adenoviral helper functions) in cell factories or multi-layer flasks to achieve 70-80% confluency at transfection.
- Plasmid Transfection: Co-transfect cells using PEI-Max with three plasmids:
- Rep/Cap Plasmid: Encodes AAV replication (Rep) and capsid (Cap) proteins. The Cap serotype dictates tropism.
- ITR-flanked Transgene Plasmid: Contains the gene of interest flanked by AAV2 inverted terminal repeats (ITRs), the only viral cis elements required for packaging.
- Adenoviral Helper Plasmid: Provides essential adenoviral genes (E2A, E4, VA RNA) for AAV replication.
- Harvest: 48-72 hours post-transfection, harvest cells and media.
- Lysis & Clarification: Freeze-thaw lysate, treat with Benzonase to degrade unpackaged nucleic acid, and clarify by centrifugation.
- Purification: Purify vector via iodixanol density gradient centrifugation or affinity chromatography (e.g., AVB Sepharose).
- Concentration & Buffer Exchange: Concentrate using centrifugal filter units and exchange into final formulation buffer (e.g., PBS + 0.001% Pluronic F-68).
- Titration: Quantify vector genome (VG) titer via qPCR against a standard curve and assess purity by SDS-PAGE/silver stain.
Protocol: Production of VSV-G Pseudotyped Lentivirus
VSV-G glycoprotein confers broad tropism and enhances vector stability.
- Cell Seeding: Seed HEK293T cells (highly transferable) in 10cm dishes.
- Plasmid Transfection: Co-transfect using calcium phosphate or PEI with four plasmids (third-generation split-genome system):
- Transfer Plasmid: Contains Ψ packaging signal, RRE, cPPT, and the transgene expression cassette flanked by LTRs.
- Packaging Plasmid (psPAX2): Provides Gag, Pol, Tat, and Rev.
- Envelope Plasmid (pMD2.G): Expresses the VSV-G glycoprotein.
- (Optional) Rev-expression Plasmid if transfer plasmid lacks a Rev-responsive element.
- Media Change: Replace media 6-8 hours post-transfection to reduce toxicity.
- Virus Harvest: Collect supernatant at 48 and 72 hours. Pool harvests, filter through a 0.45µm PES filter.
- Concentration: Concentrate vector by ultracentrifugation (e.g., 50,000 x g for 2 hours at 4°C) or using tangential flow filtration.
- Titration: Titrate on permissive cells (e.g., HEK293T) by measuring transducing units (TU/mL) via flow cytometry for a reporter gene (e.g., GFP) or by qPCR for vector copy number in transduced cells.
Visualizing Vector Lifecycles & Workflows
Diagram 1: AAV Recombinant Vector Production Workflow
Diagram 2: Lentiviral Integration Pathway
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Research Reagents for Viral Vector R&D
| Reagent/Material | Primary Function | Key Consideration |
|---|---|---|
| HEK293/HEK293T Cells | Production cell line for AAV, LV, RV, AdV. Provides necessary transcriptional milieu and helper functions. | Maintain low passage number; test for mycoplasma regularly. |
| Polyethylenimine (PEI-Max) | Cationic polymer for transient plasmid transfection. Cost-effective for large-scale production. | pH and molecular weight are critical for efficiency and toxicity. |
| Calcium Phosphate | Chemical transfection method, traditional for lentivirus production. | Precipitation condition (pH, timing) is highly sensitive for optimal results. |
| Benzonase Nuclease | Degrades unpackaged DNA/RNA in lysates, reducing viscosity and improving purity. | Essential for reducing background in qPCR titering and in vivo safety. |
| Iodixanol (OptiPrep) | Inert density gradient medium for high-purity AAV isolation via ultracentrifugation. | Separates full (infectious) particles from empty capsids based on density. |
| Polybrene (Hexadimethrine bromide) | Cationic polymer that neutralizes charge repulsion, enhancing viral attachment in vitro. | Can be cytotoxic; optimize concentration (typically 4-8 µg/mL). |
| VSV-G Envelope Plasmid (pMD2.G) | Provides broad tropism pseudotype for lentivirus, allowing concentration by ultracentrifugation. | Cytotoxic; expression must be transient. |
| qPCR Kit for ITR/ψ Region | Absolute quantification of vector genome (VG) or transducing unit (TU) titers. | Requires an appropriate standard curve (linearized plasmid). ITR-specific primers are challenging to design. |
| pAAV2/9 or pAAV2/rh10 Rep/Cap Plasmid | Provides serotype-specific capsids for AAV production, dictating in vivo tropism (e.g., CNS, liver, muscle). | Serotype choice is the single most important factor for in vivo targeting. |
| Third-Generation Lentiviral Packaging Mix (e.g., psPAX2) | Split-genome system for safer LV production, minimizing risk of RCL. | Preferable to second-gen for reduced homologuous sequence overlap. |
Within the broader thesis on the basic principles of viral and non-viral gene delivery vectors, this guide provides a technical examination of three primary non-viral archetypes. Non-viral vectors are essential for overcoming immunogenicity, insertional mutagenesis, and manufacturing scalability limitations inherent to viral platforms. This document details the core mechanisms, formulations, and experimental methodologies for Lipid Nanoparticles (LNPs), polymeric vectors, and physical delivery methods.
Lipid Nanoparticles (LNPs)
LNPs are the most clinically advanced non-viral vector, exemplified by their success in mRNA COVID-19 vaccines. They are multicomponent, self-assembled systems typically comprising four lipid types.
Core Composition & Mechanism
LNPs function by encapsulating nucleic acids, protecting them from degradation, and facilitating cellular uptake and endosomal escape. The standard four-component system includes:
- Ionizable Cationic Lipid: Forms a charge-neutral complex with nucleic acids at acidic pH during formulation, becomes positively charged in endosomes to promote membrane destabilization and escape. (e.g., DLin-MC3-DMA, SM-102, ALC-0315).
- Phospholipid: Acts as a structural lipid, contributing to bilayer formation and fusion. (e.g., DSPC).
- Cholesterol: Stabilizes the bilayer, enhances particle integrity, and modulates fluidity.
- PEGylated Lipid: Shields the particle surface, reduces aggregation, increases circulation time, and can be tuned to control cellular targeting.
Key Signaling Pathways in LNP-Mediated Delivery
The intracellular delivery of nucleic acids by LNPs involves specific pathways, particularly for endosomal escape and immune activation for mRNA vaccines.
Standard Microfluidic Formulation Protocol
Aim: To produce monodisperse, stable LNPs encapsulating mRNA or pDNA. Materials: Microfluidic mixer (e.g., NanoAssemblr, staggered herringbone design), syringes, syringe pump, lipids in ethanol, nucleic acid in acidic aqueous buffer (e.g., citrate, pH 4.0). Method:
- Prepare Lipid Stock: Mix ionizable lipid, phospholipid, cholesterol, and PEG-lipid in ethanol at a defined molar ratio (e.g., 50:10:38.5:1.5). Total lipid concentration typically 6-12 mM.
- Prepare Aqueous Phase: Dilute nucleic acid (mRNA or pDNA) in 25-50 mM citrate buffer, pH 4.0, to a target concentration (e.g., 0.1 mg/mL).
- Mixing: Load the lipid-ethanol solution and the aqueous nucleic acid solution into separate syringes. Connect syringes to the microfluidic chip.
- Formulation: Simultaneously inject both phases at a defined Total Flow Rate (TFR) and Flow Rate Ratio (FRR, typically 3:1 aqueous:ethanol). Rapid mixing induces nanoparticle self-assembly. Common parameters: TFR = 12 mL/min, FRR = 3:1.
- Buffer Exchange & Dialysis: Immediately dilute the formed LNP mixture in 1x PBS (pH 7.4) to stabilize particles. Dialyze against PBS (pH 7.4) for 4-18 hours at 4°C using a dialysis membrane (e.g., MWCO 3.5-14 kDa) to remove ethanol and adjust pH.
- Characterization: Measure particle size (Z-average, PDI) via Dynamic Light Scattering (DLS), zeta potential via Electrophoretic Light Scattering, and encapsulation efficiency using Ribogreen or PicoGreen assays.
Polymeric Vectors
Polymers condense nucleic acids into polyplexes via electrostatic interactions. Key archetypes include polyethylenimine (PEI), poly(L-lysine) (PLL), and newer biodegradable polymers.
Core Polymers & Properties
| Polymer | Structure | Mechanism of Action | Key Advantage | Key Limitation |
|---|---|---|---|---|
| Branched PEI (bPEI) | High-density amine groups | Proton-sponge effect for endosomal escape | High transfection efficiency | High cytotoxicity |
| Linear PEI (lPEI) | Linear chain of amine groups | Proton-sponge effect | More efficient & less toxic than bPEI | Still exhibits cytotoxicity |
| Poly(L-lysine) (PLL) | Primary amine backbone | Condenses DNA effectively | Biodegradable, low immunogenicity | Poor endosomal escape, low efficiency |
| Chitosan | Natural polysaccharide | Mucoadhesive, biocompatible | Excellent biocompatibility & biodegradability | Low solubility at neutral pH, moderate efficiency |
| Poly(β-amino esters) (PBAEs) | Synthesized libraries | Endosomal disruption via pH-sensitive backbone | Highly tunable, degradable, low toxicity | Requires polymer synthesis expertise |
Polyplex Formation & Transfection Workflow
The process from polymer-nucleic acid complexation to gene expression follows a defined experimental pathway.
Protocol: Polyplex Formation &In VitroTransfection
Aim: To form stable polyplexes and assess transfection efficiency in adherent cells. Materials: Polymer stock solution (e.g., 1 mg/mL PEI in water, pH 7.0), plasmid DNA (pDNA) encoding reporter gene (e.g., GFP, Luciferase), serum-free cell culture medium, HEK293 or HeLa cells. Method:
- Calculate N/P Ratio: Determine the volume of polymer needed based on the Nitrogen-to-Phosphate (N/P) ratio. For PEI, N/P ratios of 5-10 are typical. (N moles from polymer amine groups / P moles from DNA phosphate groups).
- Dilution: Dilute the required amount of pDNA in 50 µL of serum-free medium (Tube A). Dilute the calculated amount of polymer in 50 µL of the same medium (Tube B).
- Complexation: Rapidly mix Tube B (polymer) into Tube A (DNA). Vortex briefly or pipette mix.
- Incubation: Incubate the mixture at room temperature for 15-30 minutes to allow polyplex formation.
- Cell Transfection: Aspirate medium from cells seeded in a 24-well plate (70-90% confluent). Wash with PBS. Add 400 µL fresh serum-free medium to cells. Add the 100 µL polyplex mixture dropwise onto the medium. Gently swirl the plate.
- Incubation & Analysis: Incubate cells with polyplexes for 4-6 hours at 37°C. Replace medium with complete growth medium (with serum). Assay for reporter gene expression (e.g., fluorescence microscopy for GFP, luciferase assay) 24-72 hours post-transfection.
Physical Methods
Physical methods use mechanical, electrical, or hydrodynamic force to transiently permeabilize the cell membrane, allowing nucleic acids to enter directly.
Comparative Analysis of Physical Delivery Methods
| Method | Principle | Typical Application In Vitro | Typical Application In Vivo | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Electroporation | Electric pulses create membrane pores | Hard-to-transfect cells (primary cells, T-cells) | Muscle, skin, tumor (ex vivo & in vivo) | High efficiency for many cell types | High cell mortality, requires optimization |
| Microinjection | Mechanical injection via fine needle | Single-cell delivery (oocytes, embryos) | Limited (embryonic work) | Precise dosing, 100% delivery efficiency | Low throughput, technically demanding |
| Gene Gun | Ballistic delivery using gold particles | Plant cells, skin epidermal cells | Skin (DNA vaccines), mucosal surfaces | Direct delivery, no need for carriers | Tissue damage, shallow penetration |
| Sonoporation | Ultrasound with microbubbles causes cavitation | Various cell lines in culture | Tumors, heart, kidney | Non-invasive, can be targeted | Lower efficiency, requires contrast agents |
| Hydrodynamic Injection | Rapid volume injection generates pressure | N/A (in vivo only) | Mouse liver (high efficiency) | Extremely high hepatic transfection | Species/site-specific, highly invasive |
Experimental Protocol: Ex Vivo mRNA Delivery to T-cells via Electroporation
Aim: To efficiently deliver mRNA (e.g., encoding a CAR) to primary human T-cells for adoptive cell therapy research. Materials: Primary human T-cells, mRNA (IVT, purified), Electroporator (e.g., Lonza 4D-Nucleofector), appropriate electroporation cuvettes/kits (e.g., P3 Primary Cell Kit), pre-warmed culture medium. Method:
- Cell Preparation: Isolate and activate T-cells. Count and centrifuge required cells (e.g., 1-2 x 10^6). Aspirate supernatant completely.
- Sample Preparation: Resuspend cell pellet in 20 µL of room-temperature electroporation buffer from the kit. Add 2-5 µg of mRNA. Mix gently.
- Electroporation: Transfer the cell-mRNA mixture into a certified electroporation cuvette. Place cuvette in the nucleofector and run the pre-optimized program (e.g., EH-115 for human T-cells).
- Recovery: Immediately after pulsing, add 500 µL of pre-warmed (37°C) complete medium to the cuvette. Using the provided pipette, gently transfer the cells to a pre-warmed culture plate.
- Culture & Analysis: Incubate cells at 37°C. Assess cell viability (e.g., Trypan Blue) at 12-24 hours. Analyze protein expression (e.g., via flow cytometry for surface CAR) from 18 hours onwards.
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Vendor Examples (Illustrative) | Function in Non-Viral Gene Delivery Research |
|---|---|---|
| Ionizable Lipids (e.g., SM-102) | Avanti Polar Lipids, Cayman Chemical | Core functional lipid for LNP formulation; enables encapsulation and endosomal escape. |
| PEG-DMG (ALC-0159) | Avanti Polar Lipids | PEG-lipid used in LNP formulations to modulate pharmacokinetics and particle stability. |
| Polyethylenimine (PEI), linear | Polysciences, Sigma-Aldrich | Gold-standard polymer for polyplex formation; high transfection efficiency via "proton sponge". |
| Poly(β-amino ester) Libraries | Specific Synthesized (e.g., from Akinc et al. protocols) | Tunable, biodegradable polymers for screening optimal vectors for specific cell types. |
| NanoAssemblr Microfluidic Instrument | Precision NanoSystems | Enables reproducible, scalable, and tunable production of LNPs and other nanoparticles. |
| RiboGreen / PicoGreen Assay Kits | Thermo Fisher Scientific | Fluorescent nucleic acid stains for quantifying encapsulation efficiency in LNPs/polyplexes. |
| Lonza 4D-Nucleofector System | Lonza | Electroporation device for high-efficiency transfection of hard-to-transfect primary cells. |
| In Vivo-JetPEI | Polyplus-transfection | Clinically-oriented polymer transfection reagent optimized for systemic or local delivery in animals. |
| Luciferase Reporter Plasmid (pGL4) | Promega | Standardized plasmid for quantifying transfection efficiency via bioluminescence assays. |
| mScript mRNA Production System | Thermo Fisher Scientific | Complete system for in vitro transcription to produce research-grade capped/polyadenylated mRNA. |
Within the broader thesis on basic principles of viral and non-viral gene delivery vectors, three primary biological barriers consistently define therapeutic efficacy: efficient cellular uptake, successful endosomal escape, and definitive nuclear entry. This whitepaper provides an in-depth technical analysis of these hurdles, detailing the mechanisms, quantitative benchmarks, and experimental methodologies central to contemporary gene delivery research.
Cellular Uptake: Mechanisms and Quantification
Cellular uptake is the critical first step, wherein delivery vectors must navigate the plasma membrane. Mechanisms differ fundamentally between viral and non-viral systems.
Primary Uptake Pathways:
- Clathrin-Mediated Endocytosis (CME): The dominant pathway for many ligands and viral vectors (e.g., Adenovirus, AAV). It is a high-capacity, dynamin-dependent process.
- Caveolae-Mediated Endocytosis: A dynamin-dependent pathway associated with lipid rafts, often used by nanoparticles and some viruses (e.g., SV40). Leads to caveosomes.
- Macropinocytosis: A dynamin-independent, actin-driven process of engulfing large volumes of fluid and particles, exploited by larger complexes and viruses (e.g., HSV, vaccinia).
- Direct Fusion/Translocation: Specific to viral vectors (e.g., HIV-1) with fusion proteins that merge the viral envelope with the plasma membrane.
Quantitative Data on Uptake Efficiency:
Table 1: Comparative Uptake Efficiency of Select Vectors
| Vector Type | Typical Uptake Efficiency (In Vitro) | Primary Pathway(s) | Key Influencing Factor(s) |
|---|---|---|---|
| Adenovirus | >90% (in permissive cells) | CME, Macropinocytosis | CAR receptor density |
| AAV | 50-80% (serotype-dependent) | CME, Caveolae | Primary receptor (e.g., AAVR) |
| Lentivirus | 60-85% | Direct Fusion (pseudotype-dependent) | VSV-G protein & membrane lipids |
| Lipid Nanoparticles (LNPs) | 70-95% (formulation-dependent) | CME, Macropinocytosis | PEG-lipid content, ionizable lipid pKa |
| Polyethylenimine (PEI) Polyplexes | 50-90% (N/P ratio dependent) | CME, Caveolae | Particle size, surface charge (zeta potential) |
Experimental Protocol: Quantifying Uptake via Flow Cytometry
- Objective: Measure the percentage of cells that have internalized a fluorescently labeled vector.
- Materials: Cells, fluorescent vector (e.g., Cy5-labeled LNP), flow cytometer, trypsin, acid wash buffer (0.5 M NaCl, 0.2 M acetic acid, pH 2.5).
- Method:
- Seed cells in a 12-well plate and incubate to ~70% confluency.
- Incubate with the fluorescent vector at the desired concentration and time (e.g., 37°C, 4 hours).
- Critical Step: To distinguish internalized from surface-bound vector, treat cells with cold acid wash buffer for 1 minute, followed by PBS wash. This quenches extracellular fluorescence.
- Harvest cells with trypsin, wash, and resuspend in PBS containing a viability dye.
- Analyze samples using a flow cytometer. Gate on live, single cells and measure the fluorescence intensity in the appropriate channel (e.g., Cy5: 650/670 nm).
- Data Analysis: Uptake efficiency is reported as the percentage of fluorescent-positive cells and/or the geometric mean fluorescence intensity (MFI). Compare to untreated controls.
Endosomal Escape: The Critical Bottleneck
Following endocytosis, vectors are trapped within endosomal vesicles. Escape into the cytosol is necessary to avoid lysosomal degradation. This is a major point of failure for non-viral systems.
Mechanisms of Escape:
- The Proton Sponge Effect: Characteristic of polymers like PEI and PAMAM dendrimers. Their buffering capacity leads to proton and chloride influx, osmotic swelling, and endosomal rupture.
- Membrane Fusion/Disruption: Employed by viral fusion proteins (e.g., influenza HA protein, VSV-G) and synthetic fusogenic lipids (e.g., DOPE). They undergo conformational changes to merge with or disrupt the endosomal membrane.
- Pore Formation: Certain peptides (e.g., GALA, INF-7) adopt alpha-helical structures at low pH, inserting into and forming pores in the endosomal membrane.
Quantitative Data on Escape Efficiency:
Table 2: Endosomal Escape Efficiency and Strategies
| Vector/Strategy | Estimated Escape Efficiency | Mechanism | Key Limitation |
|---|---|---|---|
| Adenovirus | High (~60-80%) | Penton base protein mediates endosome lysis | Immunogenicity |
| Influenza Virus | High | HA protein low-pH triggered fusion | |
| PEI (25 kDa) | Moderate (15-30%) | Proton Sponge Effect | High cytotoxicity |
| LNPs (ionizable) | Moderate-High (20-50%) | Membrane fusion/disruption | Lipid-dependent |
| Fusogenic Peptides | Variable (10-40%) | Pore formation or membrane fusion | Serum instability, cost |
Experimental Protocol: Visualizing Endosomal Escape via Confocal Microscopy
- Objective: Co-localize a fluorescent vector with endosomal markers to confirm escape (loss of co-localization).
- Materials: Cells, fluorescent vector (e.g., Alexa Fluor 488-labeled), LysoTracker Red (for acidic compartments), Hoechst stain (nucleus), confocal microscope, live-cell imaging chamber.
- Method:
- Seed cells on glass-bottom imaging dishes.
- Pre-stain acidic endo/lysosomal compartments with LysoTracker Red (e.g., 50 nM, 30 min).
- Wash and add the fluorescent vector. Incubate for the desired time (e.g., 1-6 hours).
- Wash, add fresh media with Hoechst stain, and immediately image using a confocal microscope with appropriate laser lines and emission filters.
- Image Analysis: Use software (e.g., ImageJ, Imaris) to calculate Pearson's or Mander's co-localization coefficients between the vector (green) and LysoTracker (red) signals over time. A decrease in co-localization coefficient indicates endosomal escape.
Nuclear Entry: The Final Frontier
For DNA-based therapies, translocation across the nuclear envelope is the ultimate barrier, particularly in non-dividing cells.
Mechanisms of Nuclear Entry:
- Nuclear Pore Complex (NPC) Transport: The primary gateway. Vectors less than ~40 kDa can passively diffuse. Larger cargoes require Nuclear Localization Signals (NLS) that bind to importin α/β for active transport.
- Nuclear Envelope Breakdown (NEBD): Exploited during cell division. This is the principal mechanism for many non-viral vectors in rapidly dividing cells.
- Integrase/Transposase Activity: Used by retroviruses (HIV integrase) and transposon systems (Sleeping Beauty). They actively mediate the integration of genetic material into the host genome.
Quantitative Data on Nuclear Entry:
Table 3: Nuclear Entry Capabilities of Gene Vectors
| Vector/System | Nuclear Entry Strategy | Efficiency in Non-Dividing Cells | Key Dependency |
|---|---|---|---|
| Adenovirus | NPC disruption via hexon protein, NLS on protein VII | High | CRM1-independent import |
| AAV | Passive diffusion of ssDNA, active import via NLS on capsid | Moderate | Capsid serotype, wait for 2nd strand synthesis |
| HIV-1 (Lentivirus) | Active import via NLS in integrase & matrix proteins, PIC | High | Importin-α/β, mitosis not required |
| Non-viral NLS-Polyplex | Active import via NLS-Importin interaction | Very Low (<1%) | NLS accessibility, cargo size |
| Sleeping Beauty Transposon | Relies on mitosis (NEBD) for genomic access | Very Low in quiescent cells | Cell cycle status |
Experimental Protocol: Assessing Nuclear Import via Fractionation & qPCR
- Objective: Quantify the amount of delivered DNA that reaches the nucleus.
- Materials: Cells, DNA vector, Nuclei EZ Prep Nuclei Isolation Kit (Sigma), qPCR system, primers for the transgene, lysis buffers.
- Method:
- Treat cells with the DNA vector for the desired time (e.g., 24-48 hours).
- Wash and harvest cells. Split the sample: one for total DNA, one for nuclear fraction.
- Nuclear Fractionation: Lyse cells in hypotonic buffer with detergent (e.g., NP-40) to remove the cytoplasm. Pellet nuclei by gentle centrifugation. Wash the nuclear pellet thoroughly.
- DNA Extraction: Isolve DNA from both the total cell sample and the nuclear pellet using a DNA extraction kit.
- qPCR Analysis: Perform qPCR on both samples using primers specific to the delivered transgene. Use a single-copy endogenous gene (e.g., RNase P) to normalize for DNA input and nuclear fraction purity.
- Data Analysis: Calculate the Nuclear Delivery Efficiency as: (Transgene copy number in nuclear fraction / Transgene copy number in total cell fraction) x 100%.
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for Studying Gene Delivery Hurdles
| Reagent | Category/Example | Primary Function in Research |
|---|---|---|
| Chlorpromazine | Pharmacological Inhibitor | Inhibits clathrin-mediated endocytosis. Used to confirm uptake pathway. |
| Genistein | Pharmacological Inhibitor | Inhibits caveolae-mediated endocytosis. |
| EIPA (5-(N-ethyl-N-isopropyl)amiloride) | Pharmacological Inhibitor | Inhibits macropinocytosis. |
| LysoTracker Dyes | Fluorescent Probe (e.g., LysoTracker Red DND-99) | Stains acidic compartments (late endosomes, lysosomes). Used for co-localization studies. |
| Bafilomycin A1 | Pharmacological Inhibitor | V-ATPase inhibitor that neutralizes endosomal pH. Used to probe proton sponge effect. |
| DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine) | Helper Lipid | Fusogenic lipid used in LNPs to promote endosomal escape. |
| Nuclear Localization Signal (NLS) Peptides | Functional Peptide (e.g., SV40 Large T-antigen NLS: PKKKRKV) | Conjugated to non-viral vectors to facilitate nuclear import via importin proteins. |
| Importin-α/β Recombinant Proteins | Protein | Used in in vitro pulldown assays to validate NLS functionality and binding. |
| Digitonin | Detergent | Selective permeabilization of the plasma membrane for fractionation studies, leaving nuclear membrane intact. |
| Cell Cycle Synchronization Agents | Chemical (e.g., Thymidine, Nocodazole) | Arrest cells at specific cell cycle phases (e.g., G1/S, mitosis) to study the impact of NEBD on nuclear delivery. |
This whitepaper provides an in-depth technical examination of three critical safety barriers in gene therapy vector development: immunogenicity, insertional mutagenesis, and general toxicity. Framed within the broader thesis of basic principles governing viral and non-viral gene delivery research, this analysis underscores that overcoming these challenges is paramount for translating preclinical success into safe, effective clinical therapies. The continuous evolution of vector engineering directly targets these safety profiles, aiming to balance therapeutic potency with patient risk.
Immunogenicity: Innate and Adaptive Immune Responses
Immunogenicity refers to the ability of a vector or its transgene product to provoke an unwanted immune response. This can neutralize the therapy, reduce its durability, and cause severe adverse events.
2.1 Mechanisms & Key Players
- Innate Response: Pattern recognition receptors (PRRs) detect vector-associated molecular patterns (e.g., viral capsid, bacterial plasmid DNA, dsRNA). This triggers inflammatory cytokine release (IFN-α/β, TNF-α, IL-6) and activates antigen-presenting cells (APCs).
- Adaptive Response: APCs present vector/transgene antigens to T cells, leading to cytotoxic T lymphocyte (CTL)-mediated destruction of transduced cells and B cell-mediated neutralizing antibody (NAb) production.
2.2 Current Data Summary (2023-2024)
Table 1: Comparative Immunogenicity Profiles of Select Vectors
| Vector Type | Primary Immunogenic Component | Neutralizing Antibody Prevalence (Human) | Key Cytokine Elevation (Preclinical) | Mitigation Strategies in Development |
|---|---|---|---|---|
| AAV (Adeno-Associated Virus) | Capsid proteins, vector genome | 30-70% seroprevalence varies by serotype | IL-6, MCP-1, IFN-γ | Capsid engineering (e.g., xenotyping), Empty capsid removal, Immunosuppressive regimens |
| Adenovirus (e.g., HAdV5) | Capsid hexon, fiber proteins | ~70% global seroprevalence | High levels of IL-6, TNF-α | Rare serotype switching, Hexon chimerism, PEGylation |
| Lentivirus (LV) | Viral envelope glycoproteins | Low for VSV-G pseudotype | IFN-β, IRF3 pathway activation | Pseudotyping (e.g., with Rabies-G), Producer cell line optimization |
| LNPs (Lipid Nanoparticles) | Ionizable lipids, PEG-lipids | Anti-PEG antibodies common | Complement activation, IFN-I via cGAS-STING (if mRNA) | Novel ionizable lipids (e.g., SM-102), PEG-lipid structure optimization |
2.3 Experimental Protocol: Measuring Cell-Mediated Immune Response to Vector Antigens
- Objective: Quantify antigen-specific T cell activation following vector exposure in vivo or in vitro.
- Materials: ELISpot kit (IFN-γ), splenocytes or PBMCs from treated subjects, peptide pools spanning the immunogenic protein (e.g., capsid peptides), control peptides, cell culture medium.
- Procedure:
- Isolate PBMCs/splenocytes 7-14 days post-vector administration.
- Plate cells in ELISpot plates pre-coated with IFN-γ capture antibody.
- Stimulate with overlapping peptide pools (1-2 μg/mL per peptide) or vector particles (MOI 1000-10,000).
- Include positive (ConA/PMA) and negative (media only) controls.
- Incubate 24-48h at 37°C, 5% CO₂.
- Develop spots per manufacturer's instructions using biotinylated detection antibody, streptavidin-ALP, and BCIP/NBT substrate.
- Enumerate spots using an automated ELISpot reader. Results expressed as spot-forming cells (SFC) per million cells.
Title: Immune Activation Pathways Against Gene Therapy Vectors
Insertional Mutagenesis: Genomic Integration Risks
Insertional mutagenesis occurs when vector integration disrupts or dysregulates a host gene, potentially leading to clonal expansion and malignancy.
3.1 Mechanisms
- Oncogene Activation: Integration near a proto-oncogene promoter/enhancer leads to its overexpression.
- Tumor Suppressor Disruption: Integration disrupts the coding sequence or regulatory region of a tumor suppressor gene.
- Splicing Alterations: Vector sequences create aberrant splice sites in cellular transcripts.
3.2 Current Data Summary (2023-2024)
Table 2: Integration Profiles and Genotoxic Risk of Integrating Vectors
| Vector Type | Integration Mechanism | Integration Pattern | Risk of Genotoxicity | Risk-Mitigation Strategies |
|---|---|---|---|---|
| γ-Retrovirus (gRV) | Active, requires mitosis | Strong preference for transcriptional start sites (TSS) and CpG islands. | High (e.g., SCID-X1 trials) | Self-Inactivating (SIN) designs, use of cellular or insulatory elements. |
| Lentivirus (LV) | Active, mitosis not required | Prefers active transcriptional units (gene bodies), avoids TSS. | Significantly lower than gRV | SIN designs, Chromatin-Targeting domains (e.g., Cpfl) to steer integration to safe harbors. |
| Transposons (SB, PB) | Enzyme-mediated "cut-and-paste" | Sleeping Beauty (SB): near-TAA repeats. PiggyBac (PB): more random, slight TTAA preference. | Moderate; clonal expansion observed in some models | Hyperactive but dim. transposase variants for lower exposure; protein delivery (vs. mRNA) to limit activity window. |
| AAV | Mostly episomal; rare integration | Wild-type AAV: site-specific into AAVS1 (chr19). Recombinant AAV: random, at microhomology sites, very low frequency. | Extremely low; theoretical concern in hepatocyte proliferation. | Engineering hybrid vectors for targeted integration; enhancing episomal stability. |
3.3 Experimental Protocol: Integration Site Analysis (LAM-PCR/NGS)
- Objective: Identify and map genomic locations of vector integration sites.
- Materials: Genomic DNA from transduced cells, restriction enzymes (e.g., MseI), linkers, biotinylated primer complementary to vector LTR/inverted terminal repeat (ITR), Taq polymerase, NGS library prep kit.
- Procedure:
- Digest 1-2 µg gDNA with a frequent-cutter restriction enzyme.
- Ligate a double-stranded linker cassette to the digested ends.
- Perform linear PCR using a biotinylated vector-specific primer.
- Capture amplified products using streptavidin magnetic beads.
- Perform nested PCR using a second vector primer and a linker-specific primer to enrich integration fragments.
- Purify PCR products, prepare NGS library, and sequence on an Illumina platform.
- Bioinformatics: Map sequencing reads to the human reference genome, requiring vector-genome junction and valid linker sequence. Clonal abundance is calculated from read counts.
Title: Pathways from Vector Integration to Malignancy
Toxicity Profiles: Vector-Specific Adverse Effects
Toxicity extends beyond immune reactions to include direct pathological effects from vector components or transgene overexpression.
4.1 Key Toxicity Mechanisms
- Dose-Dependent Organ Toxicity: High vector doses can overload cellular machinery (e.g., proteasome), causing stress and apoptosis.
- Transgene-Specific Toxicity: Overexpression of a therapeutic protein can have off-target effects (e.g., coagulation factor imbalance).
- Vector Component Toxicity: Lipids in LNPs can cause hepatic or inflammatory reactions; viral capsid proteins can have direct cytotoxic effects.
- Pre-existing Immunity: As discussed in Section 2, can lead to rapid clearance and inflammatory toxicity.
4.2 Current Data Summary (2023-2024)
Table 3: Selected Toxicity Concerns and Monitoring Parameters
| Vector/Platform | Primary Target Organ | Key Toxicity Manifestation | Clinical Monitoring Parameters | Mitigation Approach |
|---|---|---|---|---|
| Systemic AAV | Liver | Hepatotoxicity (elevated transaminases), Thrombocytopenia | ALT, AST, Platelet count, Anti-capsid NAbs | Corticosteroid prophylaxis, Dose escalation studies, Alternate serotypes. |
| Systemic Adenovirus | Liver, Spleen | Acute inflammatory syndrome, Hepatotoxicity, Coagulopathy | CRP, IL-6, D-dimer, LFTs | Low-dose regimens, Potent anti-inflammatory premedication. |
| LNPs (Systemic) | Liver, Spleen | Complement activation-related pseudoallergy (CARPA), Hepatotoxicity | C3a, C5a, Bb fragment, Vital signs during infusion | Slow infusion rates, Pre-medication (antihistamines, steroids). |
| CNS-Directed Vectors | Brain | Neuroinflammation, Seizures | CSF cell count, Cytokines, MRI, EEG | Dose optimization in CSF space, Promoter engineering for cell-specificity. |
4.3 Experimental Protocol: Assessing Hepatotoxicity In Vivo
- Objective: Evaluate liver damage after systemic vector administration in a murine model.
- Materials: C57BL/6 mice, test vector, PBS control, isoflurane anesthesia, blood collection tubes, serum separator tubes, clinical chemistry analyzer.
- Procedure:
- Randomize mice into treatment (vector) and control (PBS) groups (n≥5).
- Administer vector via tail vein injection at predetermined dose(s).
- At 48h, 7d, and 28d post-injection, collect blood via retro-orbital or submandibular bleed under anesthesia.
- Allow blood to clot, centrifuge to isolate serum.
- Analyze serum for key markers: Alanine Aminotransferase (ALT) and Aspartate Aminotransferase (AST) for hepatocyte damage; Alkaline Phosphatase (ALP) for cholestasis.
- Euthanize animals at terminal time point, harvest livers for histopathology (H&E staining) and vector biodistribution analysis (qPCR for vector genomes).
- Compare treatment groups to controls using appropriate statistical tests (e.g., t-test, ANOVA).
The Scientist's Toolkit: Essential Reagents & Materials
Table 4: Key Research Reagent Solutions for Safety Assessment
| Reagent / Material | Function in Safety Research | Example / Note |
|---|---|---|
| Anti-AAV Neutralizing Antibody Assay Kit | Quantifies pre-existing or therapy-induced NAbs against specific AAV serotypes in serum/plasma. | Essential for patient screening and immunogenicity monitoring. |
| cGAS (cyclic GMP-AMP Synthase) Inhibitor | Pharmacologically inhibits the innate DNA-sensing cGAS-STING pathway. | Used in vitro to dissect mechanism of LNP or dsDNA vector immunogenicity. |
| LINE-1 (ORF2p) Antibody | Detects endogenous reverse transcriptase activity often triggered by nucleic acid vectors. | Monitors a potential mechanism of genomic instability and inflammation. |
| Humanized Mouse Models (e.g., HIS, HIL) | Engrafted with human immune system and/or liver cells. | Gold-standard for predicting human-specific immune and toxicity responses preclinically. |
| Digital Droplet PCR (ddPCR) | Absolute quantification of vector copy number per genome and detection of rare integration events. | More precise than qPCR for biodistribution and integration frequency studies. |
| Multiplex Cytokine Panels (Luminex/MSD) | Simultaneously quantifies dozens of cytokines/chemokines from small sample volumes. | Profiles immune response and inflammatory toxicity signatures. |
| Sleeping Beauty Transposon System | Tool for studying insertional mutagenesis screens and comparing integration profiles. | Used to identify common integration sites and cancer driver genes in model systems. |
| Next-Generation Sequencing (NGS) Services | For integration site analysis (LAM-PCR), off-target editing, and transcriptomic profiling. | Critical for comprehensive genomic safety assessment. |
Title: Integrated Safety Assessment Workflow for Gene Therapy Vectors
The path to safe and effective gene therapies is paved by a rigorous, multi-faceted understanding of immunogenicity, insertional mutagenesis, and toxicity. As outlined in this whitepaper, these safety pillars are intrinsically linked to the basic biological principles of vector-host interaction. Continued innovation in vector engineering—such as refined capsids, targeted integration systems, and optimized formulations—directly addresses these challenges. The experimental frameworks and tools detailed herein provide a roadmap for researchers to systematically evaluate and mitigate risks, ensuring that the foundational promise of gene therapy is realized with an unwavering commitment to patient safety.
From Bench to Bedside: Designing and Applying Gene Delivery Systems in Research
1. Introduction Within the broader investigation of gene delivery vectors, the efficient and reliable production of plasmid DNA (pDNA) is a foundational step for both non-viral transfection and the creation of viral vectors (e.g., AAV, lentivirus). This technical guide details the core experimental pipeline, from plasmid construction through to high-purity preparation, framed as a critical modular component in gene therapy and functional genomics research.
2. Plasmid Construction & Design Principles The design of the plasmid backbone dictates transgene expression, safety, and manufacturability.
- Core Elements: Essential components include an origin of replication (ori) for bacterial amplification, a selectable antibiotic resistance marker, and the eukaryotic expression cassette (promoter, transgene, poly-A signal). For viral vector production, the expression cassette is typically split across multiple plasmids (e.g., packaging, envelope, transfer plasmid) to prevent replication-competent virus formation.
- Cloning Strategy: Modern workflows favor restriction-free, sequence-independent cloning methods such as Gibson Assembly or Golden Gate Assembly, which offer high efficiency and flexibility for multi-fragment assembly.
Table 1: Common Cloning Methods - Comparison of Key Parameters
| Method | Typical Efficiency (CFU/µg) | Hands-on Time | Optimal Insert Size | Key Advantage |
|---|---|---|---|---|
| Restriction/Ligation | 1 x 10³ - 1 x 10⁴ | Moderate | 0.1 - 10 kb | Simplicity, low cost |
| Gibson Assembly | 1 x 10⁴ - 1 x 10⁶ | Low | 0.1 - 20+ kb | Seamless, multi-fragment assembly |
| Golden Gate Assembly | 1 x 10⁴ - 1 x 10⁶ | Moderate | Modular parts | Standardized, high-throughput |
| Gateway Recombination | 1 x 10⁵ - 1 x 10⁷ | Very Low | Any | Rapid, parallel cloning |
Protocol 2.1: Gibson Assembly for Vector Construction Objective: Assemble multiple DNA fragments into a linearized plasmid backbone in a single isothermal reaction. Reagents: Gibson Assembly Master Mix (commercial or prepared in-house containing T5 exonuclease, DNA polymerase, and DNA ligase), PCR-amplified insert(s), linearized vector, nuclease-free water. Procedure:
- Calculate insert:vector molar ratios. A 2:1 to 5:1 insert:vector ratio is standard. For multiple inserts, use equimolar ratios of all fragments.
- Set up a 10-20 µL reaction on ice: 5-100 ng of linearized vector, insert(s) per calculated ratio, and 1X Gibson Assembly Master Mix.
- Incubate at 50°C for 15-60 minutes.
- Transform 2-5 µL of the assembly reaction into competent E. coli cells (e.g., DH5α) via heat-shock or electroporation.
- Plate cells on LB agar with appropriate antibiotic and incubate overnight at 37°C.
- Screen colonies by colony PCR and/or restriction digest, followed by Sanger sequencing for verification.
3. Plasmid Propagation and Purification High-quality pDNA is essential for downstream applications.
Protocol 3.1: High-Purity Plasmid Preparation via Alkaline Lysis & Anion-Exchange Chromatography Objective: Isolate transfection-grade plasmid DNA from bacterial culture. Reagents: LB medium, appropriate antibiotic, P1 (Resuspension: 50 mM Tris-Cl pH 8.0, 10 mM EDTA, RNase A), P2 (Lysis: 200 mM NaOH, 1% SDS), P3 (Neutralization: 3.0 M potassium acetate, pH 5.5), endotoxin-free anion-exchange column, isopropanol, 70% ethanol, TE buffer. Procedure:
- Inoculate 50-500 mL of LB medium with a single colony and culture overnight at 37°C with shaking.
- Pellet cells by centrifugation (≥6000 x g, 15 min, 4°C). Decant supernatant completely.
- Resuspend pellet in P1 buffer (e.g., 5 mL per 50 mL culture).
- Add an equal volume of P2 buffer, mix gently by inversion, and incubate at room temperature for 3-5 min for lysis.
- Add a chilled equal volume of P3 buffer, mix immediately and gently until a white precipitate forms. Incubate on ice for 10 min.
- Centrifuge (≥15,000 x g, 30 min, 4°C) to pellet genomic DNA, proteins, and cell debris.
- Apply the cleared supernatant to an anion-exchange column. Wash with a medium-salt buffer (e.g., 0.6 M NaCl, pH ~8) to remove RNA and proteins.
- Elute pDNA with a high-salt buffer (e.g., 1.25 M NaCl, pH ~8).
- Precipitate pDNA by adding 0.7 volumes of room-temperature isopropanol, mixing, and centrifuging (≥12,000 x g, 30 min, 4°C).
- Wash the pellet with 70% ethanol, air-dry, and resuspend in TE buffer or nuclease-free water. Determine concentration and purity by absorbance (A260/A280 ratio of ~1.8-2.0) and analyze integrity by agarose gel electrophoresis.
Table 2: Plasmid Purification Methods - Yield and Quality Metrics
| Method | Scale (from Culture) | Typical Yield | A260/A280 Ratio | Endotoxin Level (EU/µg) | Recommended Use |
|---|---|---|---|---|---|
| Mini-Prep (Silica) | 1-5 mL | 5-20 µg | 1.7-1.9 | >10 | Screening, cloning |
| Maxi-Prep (Anion-Exchange) | 100-500 mL | 500-1500 µg | 1.8-2.0 | <1 | Transfection, in vitro |
| Giga-Prep (Modified Lysis/Chromatography) | 0.5-2 L | 2.5-10 mg | 1.8-2.0 | <0.1 | Viral packaging, animal studies |
| Cesium Chloride Gradient | 100-500 mL | 100-1000 µg | >1.9 | Very Low | Legacy method for highest purity |
4. Viral Vector Packaging and Purification (Lentiviral Example) This section outlines a common protocol for producing third-generation, replication-incompetent lentiviral vectors.
Protocol 4.1: Lentiviral Vector Production via Transient Transfection in HEK293T Cells Objective: Generate high-titer lentiviral particles by co-transfecting packaging and transfer plasmids. Reagents: HEK293T cells, DMEM with 10% FBS, transfection reagent (e.g., polyethylenimine (PEI)), packaging plasmids (psPAX2), envelope plasmid (pMD2.G), transfer plasmid with gene of interest, serum-free medium, 0.45 µm PVDF filter. Procedure:
- Seed HEK293T cells in a culture dish (e.g., 10 cm) to reach 70-80% confluence at the time of transfection.
- For one dish, prepare DNA mix: 10 µg transfer plasmid, 7.5 µg psPAX2, 2.5 µg pMD2.G in opti-MEM or serum-free medium (total volume ~500 µL).
- In a separate tube, dilute transfection reagent (e.g., 40 µL of 1 mg/mL PEI) in 500 µL of serum-free medium.
- Combine the DNA and PEI dilutions, mix gently, and incubate at room temperature for 15-20 min to form complexes.
- Add the complexes dropwise to the cells. Gently swirl the dish.
- After 6-8 hours, replace the medium with fresh complete medium.
- Harvest the virus-containing supernatant at 48 and 72 hours post-transfection. Pool harvests, clear cellular debris by low-speed centrifugation (500 x g, 10 min), and filter through a 0.45 µm PVDF filter. Store at 4°C short-term or at -80°C for long-term storage.
Protocol 4.2: Lentiviral Vector Concentration by Ultracentrifugation Objective: Concentrate and partially purify viral particles. Reagents: Filtered viral supernatant, Polybrene (optional), PBS, ultracentrifuge with swinging bucket rotor (e.g., SW 28). Procedure:
- Load filtered supernatant into ultracentrifuge tubes. Underlay with a 20% sucrose cushion (in PBS) if desired for higher purity.
- Centrifuge at 70,000 - 120,000 x g (e.g., 25,000 rpm in SW28 rotor) for 2 hours at 4°C.
- Carefully decant the supernatant. Invert the tube on a clean kimwipe for 5 min.
- Resuspend the often-invisible pellet in a small volume (e.g., 100-200 µL) of cold PBS or desired buffer by pipetting gently and incubating on ice for 1 hour.
- Aliquot and store at -80°C. Avoid repeated freeze-thaw cycles.
5. The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent / Material | Primary Function & Explanation |
|---|---|
| Competent E. coli Cells (e.g., DH5α, Stbl3) | Engineered for high transformation efficiency and plasmid stability; Stbl3 reduces recombination of unstable sequences (e.g., lentiviral LTRs). |
| Endotoxin-Free Plasmid Purification Kits | Utilize anion-exchange or modified silica membranes to yield pDNA with very low endotoxin levels, critical for sensitive cell culture and in vivo applications. |
| Polyethylenimine (PEI), Linear | Cationic polymer that complexes with DNA to facilitate transient transfection of adherent cells (e.g., HEK293T) for viral vector production. Cost-effective and scalable. |
| Lentiviral Packaging Mix (3rd Gen) | Pre-mixed set of plasmids (gag/pol, rev, VSV-G) for producing replication-incompetent lentivirus, improving consistency and biosafety. |
| Hexadimethrine Bromide (Polybrene) | Cationic polymer that neutralizes charge repulsion between viral particles and cell membranes, enhancing transduction efficiency. |
| Ultracentrifugation Equipment | Essential for concentrating viral vectors via high g-force pelleting. Swing-out rotors are preferred for concentrating from large volumes. |
| qPCR-based Titering Kit (Lentivirus) | Contains primers/probes for specific vector sequences (e.g., WPRE) and a standard to physically quantify vector genome copies (vg/mL), more precise than transduction assays. |
| Transduction Enhancers (e.g., Vectofusin-1) | Peptides that specifically enhance the transduction efficiency of VSV-G-pseudotyped lentivirus in difficult-to-transduce cells like primary T cells or stem cells. |
6. Visualized Workflows and Relationships
Plasmid Construction Cloning Workflow
Lentiviral Vector Production Pipeline
Plasmid Production in Gene Delivery Research
The fundamental thesis of modern gene delivery research posits that the efficacy of any genetic medicine is inextricably linked to the efficiency and specificity of its cargo delivery vector. Viral and non-viral vectors represent two divergent evolutionary paths in this field, each with distinct trade-offs between delivery efficiency, cargo capacity, immunogenicity, and manufacturability. This guide examines the core cargo-specific considerations for four pivotal therapeutic modalities—DNA, mRNA, siRNA, and CRISPR ribonucleoproteins (RNPs) or nucleic acids—within this broader vector paradigm. The choice of vector (viral: adenovirus, AAV, lentivirus; non-viral: LNPs, polymers, peptides) must be meticulously aligned with the physicochemical properties, intracellular destination, and functional requirements of the molecular cargo.
Cargo-Specific Characteristics & Delivery Challenges
Table 1: Core Characteristics and Delivery Requirements of Nucleic Acid Cargos
| Cargo Type | Size (approx.) | Charge (@ pH 7) | Primary Target | Key Delivery Challenge | Stability Requirement |
|---|---|---|---|---|---|
| Plasmid DNA | 3-20 kbp | Highly negative (polyanionic) | Nucleus (for transcription) | Nuclear envelope penetration; risk of genomic integration | High; must resist nucleases |
| mRNA | 1-5 kb | Negative | Cytoplasm (for translation) | Cytosolic RNase degradation; innate immune activation | Moderate; requires 5' cap & modified nucleotides |
| siRNA | 19-27 bp dsRNA | Negative | Cytoplasm (RISC loading) | Off-target effects; endosomal trapping; transient effect | Low; but must be duplexed for RISC entry |
| CRISPR RNP | ~160 kDa Cas9 + gRNA | Negative (gRNA) / Variable (Cas9) | Nucleus or Cytoplasm | Co-delivery of large protein & RNA; timing of function | High for protein integrity; gRNA stability |
Table 2: Common Vector Suitability by Cargo Type (2024 Landscape)
| Vector Class | DNA | mRNA | siRNA | CRISPR (plasmid) | CRISPR (mRNA+gRNA) | CRISPR (RNP) |
|---|---|---|---|---|---|---|
| Viral | ||||||
| Adenovirus | Excellent | N/A | Poor | Good | N/A | Poor |
| AAV | Good (size-limited) | N/A | Poor | Limited | N/A | Poor |
| Lentivirus | Excellent | N/A | Poor | Good (integrating) | N/A | Poor |
| Non-Viral | ||||||
| Lipid Nanoparticles (LNPs) | Good | Excellent (current standard) | Excellent (first approved) | Good | Good | Good (evolving) |
| Polymeric Nanoparticles | Good | Good | Good | Good | Good | Fair |
| Electroporation (ex vivo) | Excellent | Excellent | Excellent | Excellent | Excellent | Excellent |
Detailed Methodologies & Experimental Protocols
Protocol: Formulation of Ionizable Lipid Nanoparticles (LNPs) for mRNA Delivery
Adapted from current Good Manufacturing Practice (cGMP) guidelines for clinical-grade formulation.
Objective: To reproducibly formulate mRNA-loaded LNPs using microfluidic mixing. Materials:
- Lipid Stock Solutions: Ionizable lipid (e.g., DLin-MC3-DMA), DSPC, Cholesterol, PEG-lipid dissolved in ethanol.
- Aqueous Phase: mRNA in citrate buffer (pH 4.0).
- Equipment: Precision microfluidic mixer (e.g., NanoAssemblr), PBS (pH 7.4), dialysis cassettes (MWCO 20 kDa). Procedure:
- Prepare the lipid mixture in ethanol at a molar ratio (e.g., 50:10:38.5:1.5 ionizable lipid:DSPC:Chol:PEG-lipid) to a total concentration of 10 mg/mL.
- Prepare the mRNA solution in 50 mM citrate buffer at a concentration of 0.2 mg/mL. Maintain RNAse-free conditions.
- Set the flow rate ratio (aqueous:organic) to 3:1 on the microfluidic mixer. Typical total flow rate is 12 mL/min.
- Load the two solutions into separate syringes and initiate mixing. The collected effluent is a turbid suspension of mRNA-LNPs.
- Immediately dilute the effluent with 4x volume of PBS (pH 7.4) to allow particle maturation.
- Dialyze against PBS (pH 7.4) for 18 hours at 4°C to remove ethanol and exchange the buffer.
- Filter the final formulation through a 0.22 µm sterile filter. Characterize particle size (DLS), PDI, encapsulation efficiency (RiboGreen assay), and mRNA integrity (gel electrophoresis).
Protocol: Transfection of CRISPR-Cas9 RNP Using Electroporation (Ex Vivo)
Objective: To deliver pre-assembled Cas9 protein:gRNA ribonucleoprotein (RNP) complexes into primary T-cells for gene editing. Materials: Neon Transfection System (Thermo Fisher), Cas9 Nuclease (100 µM), synthetic gRNA (100 µM), P3 Primary Cell Nucleofector Kit, pre-activated human T-cells. Procedure:
- Pre-assemble RNP complexes by mixing Cas9 protein and gRNA at a 1:2 molar ratio in a low-salt buffer. Incubate at room temperature for 15 minutes.
- Harvest 1x10^6 activated T-cells, wash twice with PBS, and resuspend in 20 µL of Resuspension Buffer R from the Nucleofector Kit.
- Mix 20 µL cell suspension with 5 µL of pre-assembled RNP complexes (e.g., 10 pmol Cas9). Transfer mixture to a Neon tip.
- Electroporate using the pre-set program for primary human T-cells (e.g., 1700V, 20ms, 1 pulse).
- Immediately transfer electroporated cells to pre-warmed, cytokine-supplemented RPMI medium.
- Assess editing efficiency at the target locus 72 hours post-electroporation via T7 Endonuclease I assay or next-generation sequencing.
Visualizing Key Pathways and Workflows
Diagram Title: LNP-mRNA Delivery and Endosomal Escape Pathway
Diagram Title: CRISPR Delivery Workflow: Viral vs Non-Viral Paths
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Nucleic Acid Delivery Research
| Reagent / Material | Primary Function | Example Use Case | Key Consideration |
|---|---|---|---|
| Ionizable Cationic Lipids | Core component of LNPs; protonates in endosome to enable membrane disruption/mRNA release. | Formulating mRNA vaccines (e.g., SM-102, ALC-0315). | pKa must be ~6-7 for optimal endosomal escape. |
| Polyethyleneimine (PEI) | High cationic charge density polymer condenses nucleic acids; promotes endosomal escape via "proton sponge" effect. | Transfection of plasmid DNA in vitro. | High molecular weight PEI is efficient but cytotoxic. |
| Adeno-Associated Virus (AAV) Serotypes | Viral capsid proteins determining tropism (e.g., AAV9 crosses BBB, AAV6 targets lungs). | In vivo gene therapy for inherited diseases. | Pre-existing immunity limits patient population. |
| Nucleofector Kits | Cell-type specific electroporation buffers and protocols. | Ex vivo editing of hard-to-transfect primary cells (T-cells, stem cells). | Optimization of pulse parameters is critical for viability. |
| RiboGreen Assay Kit | Fluorescent dye that selectively binds RNA/DNA; measures encapsulation efficiency. | Quantifying % of nucleic acid cargo loaded inside LNPs. | Requires careful use of detergent to distinguish free vs. encapsulated cargo. |
| T7 Endonuclease I | Detects mismatches in heteroduplex DNA formed after imperfect NHEJ repair. | Initial assessment of CRISPR-Cas9 editing efficiency. | Can under-report efficiency, especially for HDR edits. |
| Nucleoside-Modified mRNA | Incorporation of pseudouridine (Ψ) or 5-methylcytidine reduces innate immune recognition by pattern recognition receptors (PRRs). | Improving translational yield and safety of mRNA therapeutics. | Must be incorporated during in vitro transcription (IVT). |
Within the broader thesis on the basic principles of gene delivery vectors, the transition from broad, systemic administration to precise, targeted delivery represents a pivotal frontier. This whitepaper provides an in-depth technical guide to two cornerstone targeting strategies: the use of tissue-specific promoters (TSPs) for transcriptional targeting and surface ligand modifications for transductional targeting. While viral vectors (e.g., AAV, lentivirus) and non-viral vectors (e.g., LNPs, polymeric nanoparticles) possess inherent tropisms, engineering them with these strategies is essential to enhance efficacy and safety by directing transgene expression to specific cell types while minimizing off-target effects and immune responses.
Transcriptional Targeting with Tissue-Specific Promoters
Transcriptional targeting employs regulatory DNA sequences (promoters, enhancers) that are selectively active in particular tissues or cell types, thereby restricting transgene expression even if the vector transduces a broader range of cells.
Core Principles and Design
A TSP is characterized by its ability to drive gene expression preferentially in a target tissue due to the presence of cell-type-specific transcription factors. Key design considerations include:
- Strength vs. Specificity: Often a trade-off exists; strong viral promoters (e.g., CMV) are active ubiquitously. TSPs are typically weaker but more precise.
- Size: Larger promoter fragments may contain necessary distal enhancer elements but pose packaging constraints, especially for AAV vectors (~4.7 kb capacity).
- Epigenetic Stability: Promoters should resist silencing over time, a challenge for some viral sequences.
Quantitative Comparison of Common Tissue-Specific Promoters
Table 1: Characteristics of Selected Tissue-Specific Promoters
| Promoter Name | Target Tissue/Cell Type | Approximate Size (bp) | Relative Strength (% of CMV) * | Key Advantages | Limitations |
|---|---|---|---|---|---|
| Synapsin I | Neurons | ~470 | ~15-30% | Highly neuron-specific; good for CNS gene therapy. | Weak activity in some neuronal subtypes. |
| Albumin | Hepatocytes | ~300 - 2000 | ~50-150% | Very strong and specific in liver; multiple enhancer regions. | Size of full element can be large. |
| Cardiac Troponin T (cTnT) | Cardiomyocytes | ~400 | ~20-40% | High specificity for heart muscle. | Moderate strength. |
| hKRT14 | Keratinocytes (Skin) | ~2100 | ~10-20% | Specific for epidermal basal layer. | Large size; relatively weak. |
| PSE (Probasin) | Prostate | ~600 | ~5-10% | Androgen-responsive; high prostate specificity. | Very weak; hormone-regulated. |
| Tie2 | Vascular Endothelium | ~300 - 1500 | ~5-15% | Endothelial-specific; used in anti-angiogenic therapy. | Often requires enhancer for robust activity. |
Strength is highly dependent on cell type and vector context; values are approximate from comparative literature. The hepatic locus control region combined with a minimal albumin promoter can exceed CMV activity in hepatocytes.
Experimental Protocol: Validating Promoter SpecificityIn VitroandIn Vivo
Aim: To assess the activity and specificity of a candidate TSP driving a reporter gene (e.g., luciferase, GFP) compared to a ubiquitous promoter.
Materials:
- Vector Construction: Recombinant AAV or lentiviral vectors encoding firefly luciferase (Fluc) under the control of the TSP and a control promoter (e.g., CMV or CAG).
- Cells: A panel of relevant cell lines (target cell line and 2-3 non-target cell lines).
- Animals: Appropriate mouse model (e.g., C57BL/6) for in vivo biodistribution.
Procedure:
In Vitro Transduction:
- Seed target and non-target cell lines in 24-well plates.
- Transduce cells with vectors at a fixed multiplicity of infection (MOI) in triplicate.
- After 48-72 hours, lyse cells and quantify luciferase activity using a luminometer (relative light units, RLU). Normalize RLU to total protein content (Bradford assay).
- Analysis: Calculate the ratio of TSP activity (RLU/mg protein) in target vs. non-target cells. Compare to the same ratio for the CMV promoter vector. A high target:non-target ratio for the TSP indicates specificity.
In Vivo Biodistribution (IVIS Imaging):
- Administer vectors systemically (e.g., intravenous) or locally to mice (n=5 per group).
- At predetermined time points (e.g., day 7, 14), inject mice with D-luciferin substrate (150 mg/kg, i.p.).
- Anesthetize mice and image using an In Vivo Imaging System (IVIS) to capture bioluminescence.
- Euthanize animals, harvest organs (target tissue, liver, spleen, heart, lung, etc.), and image ex vivo.
- Analysis: Quantify total flux (photons/sec) for each organ. Plot the biodistribution profile. Specificity is demonstrated by high signal in the target organ and low signal in off-target organs for the TSP vector, contrasting with the broad expression of the CMV vector.
Transductional Targeting via Surface Ligand Modifications
This strategy involves physically engineering the surface of the vector particle to display ligands that bind to receptors uniquely or abundantly expressed on target cells.
Quantitative Comparison of Ligand-Targeted Vector Systems
Table 2: Common Ligand-Receptor Pairs for Vector Targeting
| Ligand | Target Receptor | Primary Tissue/Cell Target | Vector Platform (Example) | Conjugation Method | Key Challenge |
|---|---|---|---|---|---|
| N-acetylgalactosamine (GalNAc) | Asialoglycoprotein Receptor (ASGPR) | Hepatocytes | siRNA-LNP, AAV, ASO | Covalent lipid conjugation, genetic fusion | Almost exclusive to hepatocytes; limited to liver. |
| RGD Peptide | αvβ3 / αvβ5 Integrins | Tumor vasculature, activated endothelium | Polymeric NPs, Adenovirus | Chemical coupling to PEG-lipid, peptide insertion in capsid | Widespread expression of integrins can reduce specificity. |
| Transferrin | Transferrin Receptor (TfR) | Brain endothelium (via transcytosis), proliferating cells | Liposomes, Gold NPs | Antibody conjugation, ligand grafting | High endogenous levels can saturate binding. |
| Anti-HER2 scFv | HER2/neu | HER2+ Breast Cancer cells | Lentivirus, Retrovirus | Genetic fusion to envelope glycoprotein (pseudotyping) | Immunogenicity of antibody fragments. |
| EGF | Epidermal Growth Factor Receptor | Epithelial cells, certain tumors | AAV, PEI Polyplexes | Biotin-streptavidin bridge, chemical conjugation | Potential for receptor-mediated downstream signaling. |
| Follicle-Stimulating Hormone (FSH) | FSHR | Ovarian granulosa cells, testicular Sertoli cells | AAV | Genetic capsid insertion (AAV library selection) | Very specialized application. |
Experimental Protocol: Conjugating Ligands to Non-Viral Lipid Nanoparticles (LNPs)
Aim: To produce and characterize LNPs decorated with a targeting ligand (e.g., GalNAc for hepatocytes).
Materials:
- Lipids: Ionizable lipid (e.g., DLin-MC3-DMA), DSPC, Cholesterol, PEG-lipid (e.g., DMG-PEG2000), and Maleimide-functionalized PEG-lipid (Mal-PEG-DSPE).
- Ligand: Thiolated GalNAc (GalNAc-SH).
- Payload: siRNA or mRNA.
- Equipment: Microfluidic mixer (e.g., NanoAssemblr), PD-10 desalting column, Zetasizer.
Procedure:
LNP Formulation (Rapid Mixing):
- Prepare an ethanol phase containing ionizable lipid, DSPC, cholesterol, and PEG-lipid (with 0.5-1 mol% of Mal-PEG-DSPE) at a total lipid concentration of ~10 mM.
- Prepare an aqueous phase containing the nucleic acid payload in citrate buffer (pH 4.0).
- Use a microfluidic mixer to combine the ethanol and aqueous phases at a fixed flow rate ratio (typically 3:1 aqueous:ethanol) and a total flow rate of ~12 mL/min. Collect the formed LNPs in a PBS buffer (pH 7.4).
Ligand Conjugation (Post-Insertion):
- Incubate the freshly prepared LNPs with thiolated GalNAc ligand (10-fold molar excess to maleimide groups) for 2-4 hours at room temperature under gentle agitation.
- Pass the reaction mixture through a PD-10 desalting column equilibrated with PBS to remove unreacted ligand and exchange the buffer.
Characterization:
- Size and Zeta Potential: Measure hydrodynamic diameter and polydispersity index (PDI) via Dynamic Light Scattering (DLS). Measure zeta potential in PBS.
- Ligand Coupling Efficiency: Quantify using a colorimetric assay for free thiol groups (e.g., Ellman's reagent) before and after conjugation, or via HPLC analysis of uncoupled ligand in the flow-through.
- In Vitro Validation: Treat ASGPR-expressing (HepG2) and non-expressing (SK-HEP-1) cells with targeted and non-targeted LNPs containing Cy5-labeled siRNA. Quantify cellular uptake by flow cytometry after 4 hours. Perform functional knockdown of a target gene (e.g., TTR) via qPCR.
Integrated Pathways and Experimental Workflow
Diagram 1: Gene Delivery Targeting Strategy Decision Logic (99 chars)
Diagram 2: Experimental Workflow for Targeted Vector Development (99 chars)
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Targeting Strategy Research
| Reagent / Material | Function in Research | Example Vendor/Catalog | Key Consideration |
|---|---|---|---|
| pAAV-MCS Expression Kit | Provides AAV ITR-containing backbone for cloning TSPs and transgenes. | Cell Biolabs, V7601 | Contains essential ITRs but lacks promoter; ideal for custom TSP insertion. |
| Lentiviral Packaging Mix (2nd/3rd Gen) | For producing lentiviral vectors with pseudotyped envelopes (e.g., VSV-G) for ligand engineering. | Takara Bio, 631275 | Envelope plasmid can be replaced with one encoding a fusion targeting ligand. |
| Ionizable Cationic Lipid (DLin-MC3-DMA) | Key component of modern LNPs for encapsulating nucleic acids. | MedChemExpress, HY-130024 | Critical for endosomal escape; proprietary analogues are common in industry. |
| Maleimide-PEG-DSPE | Functionalized lipid for post-insertion ligand conjugation to liposomes/LNPs via thiol-maleimide chemistry. | Nanocs, PG2-DSPML-5k | Maleimide group has limited stability in aqueous buffer; use fresh preparations. |
| Thiolated Targeting Ligand (e.g., GalNAc-SH) | Ready-to-conjugate ligand for surface modification of nanoparticles. | BroadPharm, BP-25681 | Ensure sufficient purity and degree of thiolation per molecule. |
| In Vivo Imaging System (IVIS) & D-Luciferin | Enables non-invasive, longitudinal tracking of bioluminescent reporter gene expression in vivo. | PerkinElmer, 122799 | Luciferase kinetics differ by tissue; standardize imaging time post-injection. |
| Cell-Type Specific Transcription Factor Antibody Panel | For validating the activity of endogenous TSP pathways via ChIP or Western Blot. | Cell Signaling Technology | Confirms the molecular basis for TSP activity in your target cell line. |
| Recombinant Target Receptor Protein (Fc-tagged) | For in vitro binding assays (ELISA, SPR) to validate ligand-receptor interaction. | R&D Systems | Use as a coating antigen to measure binding affinity of your targeted vector. |
The methodologies of transfection (non-viral gene delivery) and transduction (viral-mediated gene delivery) constitute the practical backbone of genetic research and therapeutic development. Within the broader thesis on gene delivery vectors, mastering these in vitro and in vivo techniques is not merely procedural but fundamental to understanding vector efficiency, tropism, cytotoxicity, and ultimate therapeutic potential. This guide synthesizes current standards and best practices, providing a rigorous technical framework for researchers.
Core Principles: Viral vs. Non-Viral Delivery
The choice between viral and non-viral methods hinges on the experimental goals: high efficiency and long-term expression often favor viral vectors, while safety, lower immunogenicity, and ease of scale-up can favor non-viral approaches.
- Transfection: The introduction of nucleic acids into eukaryotic cells using chemical, lipid, or physical methods. It is transient (typically) and does not involve viral particles.
- Transduction: The use of viral vectors (e.g., lentivirus, AAV, adenovirus) to deliver genetic material, which can lead to stable integration (lentivirus) or long-term episomal expression (AAV).
In VitroTransfection: Standard Protocols
Lipid-Based Transfection (Gold Standard for Many Cell Lines)
Principle: Cationic lipids complex with negatively charged nucleic acids to form lipoplexes, which fuse with the cell membrane and release cargo into the cytoplasm.
Detailed Protocol (for a 24-well plate):
- Day 0: Seed cells in 500 µL of complete growth medium without antibiotics to achieve 70-90% confluency at the time of transfection (24-48 hours later).
- Day 1 (Transfection): a. Dilution A: Dilute 0.5-1 µg of plasmid DNA in 50 µL of serum-free, antibiotic-free medium (e.g., Opti-MEM). b. Dilution B: Dilute 1-3 µL of lipid reagent (e.g., Lipofectamine 3000) in 50 µL of the same medium. Incubate for 5 minutes at room temperature. c. Complexation: Combine Dilution A and B, mix gently. Incubate for 15-20 minutes at room temperature to allow lipoplex formation. d. Addition: Add the 100 µL complex dropwise to the cells in the well. Gently rock the plate. e. Incubation: Incubate cells at 37°C, 5% CO₂ for 4-6 hours.
- Medium Change: Replace transfection mixture with 1 mL of fresh complete growth medium.
- Analysis: Assay for gene expression typically 24-72 hours post-transfection.
Lentiviral Transduction
Principle: VSV-G pseudotyped lentivirus enters cells via receptor-mediated endocytosis, leading to RNA genome reverse transcription and integration into the host genome for stable expression.
Detailed Protocol (with Polybrene):
- Day 0: Plate target cells in complete medium. Target 30-50% confluency at transduction.
- Day 1 (Transduction): a. Thaw viral supernatant on ice. Gently mix. b. Prepare dilution of viral particles in fresh medium containing polycation enhancer Polybrene (final concentration 4-8 µg/mL). Note: Multiplicity of Infection (MOI) must be determined empirically; a range of 1-10 is common. c. Remove medium from cells and add the virus/polybrene mixture. d. Centrifuge plates at 800-1000 x g for 30-60 minutes at 32°C (spinoculation). This enhances transduction efficiency. e. Incubate cells at 37°C, 5% CO₂ for 4-24 hours.
- Day 2: Remove virus-containing medium and replace with fresh complete medium.
- Days 3-5: Begin selection with appropriate antibiotic (e.g., puromycin) if vector contains a resistance gene, or analyze expression via fluorescence/assay.
Quantitative Data Comparison:In VitroMethods
Table 1: Comparison of Common In Vitro Gene Delivery Methods
| Method | Typical Efficiency (Adherent Cells) | Onset of Expression | Duration of Expression | Key Advantages | Primary Limitations |
|---|---|---|---|---|---|
| PEI Transfection | 70-90% (HEK293) | 24-48 hrs | Transient (5-7 days) | Low cost, high efficiency for many lines, works in vivo | Cytotoxic at high doses, polymer batch variability |
| Lipid Nanoparticles | 50-95% (varies) | 4-24 hrs | Transient (3-5 days) | High efficiency, low immunogenicity in vitro, good for siRNA/mRNA | Can be expensive, optimization required |
| Lentivirus | >90% (dividing cells) | 48-72 hrs | Stable/Integrating | Stable expression, broad tropism, infects dividing/non-dividing | Biosafety Level 2, insertional mutagenesis risk, size limit (~8kb) |
| Adeno-Associated Virus (AAV) | 60-80% (permissive) | 72-96 hrs | Long-term episomal | Low immunogenicity, high safety profile, strong in vivo use | Small cargo capacity (~4.7kb), pre-existing immunity |
| Electroporation | 40-80% (primary cells) | 12-24 hrs | Transient or Stable | Works for hard-to-transfect cells (e.g., primary, immune cells) | High cell mortality, requires specialized equipment |
In VivoDelivery: Best Practices and Protocols
In vivo delivery introduces significant complexity, including immune system interaction, biodistribution, and targeting.
Systemic AAV Delivery (Tail Vein Injection in Mice)
Principle: AAV serotypes (e.g., AAV8, AAV9) have specific tissue tropisms. Systemic injection leads to broad distribution, notably to liver, heart, and CNS.
Detailed Protocol:
- Viral Preparation: Thaw and keep AAV vector stock on ice. Dilute in sterile, ice-cold PBS to the desired dose (typical range: 1e10 - 1e13 vector genomes (vg) per mouse). Keep diluted virus on ice.
- Animal Preparation: Place mouse in a restraining device with tail exposed. Warm tail under a heat lamp (≤ 5 mins) to dilate veins.
- Injection: Using a 29-30G insulin syringe, slowly inject 100-200 µL of the viral solution into the lateral tail vein. A flash of blood indicates proper entry. Administer slowly over ~30 seconds.
- Post-Injection: Apply gentle pressure to the site with gauze. Return animal to cage and monitor.
- Analysis: Tissues can be harvested and analyzed for transgene expression typically 2-4 weeks post-injection to allow for maximal expression.
2In VivoJetPEI DNA Transfection (Local/Tumor Delivery)
Principle: Linear polyethylenimine (PEI) condenses DNA into positively charged nanoparticles that can be taken up by cells in situ.
Detailed Protocol (for subcutaneous tumor):
- Complex Formation: For a 100 µL injection volume, mix 10-20 µg of endotoxin-free plasmid DNA in 50 µL of 5% glucose. In a separate tube, dilute in vivo-grade JetPEI reagent in 50 µL of 5% glucose (N/P ratio typically 6-10). Combine the two solutions, vortex immediately for 10 seconds.
- Incubation: Allow complexes to form for 15-30 minutes at room temperature.
- Injection: Load complex into an insulin syringe. Inject intratumorally or subcutaneously near the target tissue in an anesthetized mouse. Multiple injections may be performed.
- Analysis: Expression is usually transient, peaking at 24-48 hours.
Quantitative Data Comparison:In VivoMethods
Table 2: Comparison of Common In Vivo Gene Delivery Routes & Vectors
| Vector/ Method | Common Route | Primary Target Tissues | Expression Onset | Duration | Key Consideration |
|---|---|---|---|---|---|
| AAV8 | Systemic (IV) | Liver, Heart, Muscle | 7-14 days | Years (episomal) | High liver tropism, moderate immunogenicity |
| AAV9 | Systemic (IV) | Heart, CNS, Pancreas | 7-14 days | Years (episomal) | Crosses blood-brain barrier efficiently |
| Lentivirus | Local (e.g., intratumoral) | Dividing cells at site | 3-7 days | Stable/Integrating | Localized delivery mitigates biosafety risks |
| Cationic Liposomes | Intravenous (IV) or Local | Lung, Spleen, Endothelium (IV) | 24-48 hrs | Transient (days) | Can activate complement system, dose-limiting toxicity |
| Hydrodynamic Injection | High-volume IV (mouse) | Hepatocytes (>90%) | 6-24 hrs | Transient (weeks) | Mouse-specific, traumatic but extremely efficient for liver |
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagent Solutions for Transfection/Transduction Workflows
| Reagent/Material | Function & Principle | Example Product/Brand |
|---|---|---|
| Polybrene (Hexadimethrine Bromide) | A polycation that reduces electrostatic repulsion between viral particles and cell membrane, enhancing viral adhesion and uptake. | Sigma-Aldrich H9268 |
| Lipofectamine 3000 | A proprietary lipid nanoparticle formulation optimized for plasmid DNA and siRNA delivery, offering high efficiency and reduced cytotoxicity. | Invitrogen L3000001 |
| Polyethylenimine (PEI), linear | A cationic polymer that condenses DNA into nanoparticles via charge interaction. The "gold standard" for transient transfection and in vivo use due to cost-effectiveness. | Polysciences 23966-2 |
| FuGENE HD | A non-liposomal lipid formulation known for low cytotoxicity and high efficiency across a wide range of cell lines, including sensitive ones. | Promega E2311 |
| Opti-MEM I Reduced Serum Medium | A low-serum, phenol-red free medium used for diluting lipids/DNA during complex formation, minimizing interference and toxicity. | Gibco 31985062 |
| Puromycin Dihydrochloride | An aminonucleoside antibiotic that inhibits protein synthesis. Used as a selection agent for cells transduced/transfected with vectors containing a puromycin resistance gene (pac). | Gibco A1113803 |
| Poly-D-Lysine | A synthetic polymer used to coat tissue culture surfaces, enhancing the attachment of adherent cells, particularly primary neurons. | Corning 354210 |
| Lenti-X Concentrator | A proprietary reagent that precipitates lentiviral particles from large volumes of supernatant, enabling high-titer viral stock production. | Takara Bio 631231 |
Critical Signaling & Cellular Pathways in Gene Delivery
Diagram Title: Cellular Uptake and Intracellular Trafficking Pathways for Viral and Non-Viral Vectors
Experimental Workflow: From Design to Analysis
Diagram Title: Generalized Workflow for Gene Delivery Experiments
The rigorous application of standardized in vitro and in vivo protocols is critical for generating reproducible, high-quality data in gene delivery research. The choice of vector and method must be aligned with the specific biological question within the broader thesis—whether it demands transient knockdown, stable overexpression, or therapeutic translation. Continuous optimization, guided by quantitative metrics and a deep understanding of intracellular trafficking pathways, remains the cornerstone of advancing the field of genetic medicine.
This whitepaper, framed within the context of a broader thesis on basic principles of viral and non-viral gene delivery vectors, presents technical case studies of three key therapeutic applications. The success of these modalities hinges on the fundamental engineering of their delivery vectors to achieve specificity, efficiency, and safety.
Case Study: Gene Therapy for Spinal Muscular Atrophy (SMA)
Therapeutic Agent: Onasemnogene abeparvovec (Zolgensma). Delivery Vector: Adeno-associated virus serotype 9 (AAV9). Principle: AAV9 is a non-enveloped, single-stranded DNA viral vector engineered to deliver a functional copy of the human SMN1 gene to motor neuron nuclei via systemic administration. Its natural tropism for crossing the blood-brain barrier and transducing neurons is leveraged for central nervous system (CNS) targeting.
Key Clinical Data: Table 1: Summary of Pivotal Clinical Trial Data for Onasemnogene Abeparvovec (STR1VE-US, NCT03306277)
| Parameter | Interim Data (Patients, n=22) | Historical Natural History |
|---|---|---|
| Event-free Survival at 14 Months | 91% (20/22) | 26% |
| Independent Sitting (≥30 seconds) at 18 Months | 59% (13/22) | 3% |
| Mean CHOP-INTEND Score Increase (from baseline) | +24.9 points at Month 12 | Progressive Decline |
| Serious Adverse Events (SAEs) | 23% (5/22) related to treatment | N/A |
| Elevated Liver Enzymes | 68% (15/22), managed with prednisolone | N/A |
Experimental Protocol (Key Preclinical Biodistribution Assay):
- Objective: Quantify vector genome copies in target and off-target tissues following intravenous administration in a murine model.
- Materials: AAV9-CBh-SMN1 vector, neonatal mice (P1), phosphate-buffered saline (PBS), dissection tools, DNA extraction kit, TaqMan qPCR reagents, tissue homogenizer.
- Procedure:
- Inject mice intravenously with 2x10^14 vg/kg of AAV9 vector or PBS (control).
- Euthanize animals at 14 days post-injection.
- Harvest tissues: spinal cord, brain, liver, heart, skeletal muscle.
- Homogenize tissues and extract total genomic DNA.
- Perform TaqMan qPCR using primers/probe specific to the vector transgene (e.g., CBh promoter) and a standard curve of vector plasmid.
- Normalize data as vector genomes (vg) per diploid genome (vg/dg) or per microgram of total DNA.
Diagram 1: AAV9 Biodistribution Assay Workflow (78 chars)
The Scientist's Toolkit: Key Reagents for AAV Gene Therapy Research
| Research Reagent | Function & Rationale |
|---|---|
| AAV Purification Kits (Iodixanol Gradient or Affinity) | Isolate and concentrate high-titer, high-purity AAV vectors from cell lysates. Affinity resins (e.g., AVB Sepharose) offer serotype-specific capture. |
| DNase I (RNase-free) | Distinguish between internalized/encapsidated vs. external/unpackaged vector genomes prior to qPCR titering. |
| TaqMan qPCR Master Mix | Precisely quantify vector genome titers (physical/genomic titer) in purified lots and biodistribution samples with high sensitivity and specificity. |
| Anti-AAV Capsid Antibodies | Characterize vector purity (SDS-PAGE/Western), perform ELISA for titer (immunocapsid titer), or detect neutralizing antibodies (NAbs) in sera. |
| Prednisolone | Standard of care immunosuppressant to mitigate potential AAV capsid-directed T-cell responses and treat vector-related transaminitis in preclinical/clinical studies. |
Case Study: mRNA Vaccine for SARS-CoV-2
Therapeutic Agent: BNT162b2 (Comirnaty) mRNA-LNP. Delivery Vector: Lipid Nanoparticle (LNP) – a non-viral vector. Principle: LNPs encapsulate and protect nucleoside-modified mRNA encoding the SARS-CoV-2 Spike glycoprotein. Upon intramuscular injection and cellular uptake, the mRNA is translated in the cytoplasm to produce the immunogenic antigen, eliciting humoral and cellular immune responses.
Key Clinical & Real-World Data: Table 2: Efficacy and Immunogenicity of BNT162b2 mRNA-LNP Vaccine (Pivotal Phase 3 Trial & Subsequent Analysis)
| Parameter | BNT162b2 Efficacy/Response | Comparator/Notes |
|---|---|---|
| Phase 3 Efficacy vs. COVID-19 | 95% (95% CI: 90.3-97.6) | Placebo, median 2-month follow-up |
| Geometric Mean Titer (GMT) of Neutralizing Antibodies | ~3.8x the GMT of convalescent sera | Assayed at 1 month post-dose 2 |
| Vaccine Effectiveness vs. Delta Variant | 88% (95% CI: 85.3-90.1) | Real-world data, symptomatic disease |
| T-cell Response (IFN-γ ELISpot) | Robust CD4+ and CD8+ T-cell responses detected | Polyfunctional Th1-skewed response |
| Local/Systemic Reactogenicity | High frequency, mild-moderate (e.g., pain, fatigue) | Generally resolved within 1-2 days |
Experimental Protocol (Key In Vitro mRNA Translation & Immunogenicity Assay):
- Objective: Evaluate mRNA-LNP potency by measuring antigen expression and subsequent immune cell activation.
- Materials: mRNA-LNP formulation, HEK-293T or dendritic cells, flow cytometer, ELISA/ECLIA kits for cytokine detection (IFN-γ, IL-2, IL-4), MHC-I tetramers (for CD8+ T-cells).
- Procedure:
- Transfect or treat antigen-presenting cells (APCs) in vitro with mRNA-LNPs.
- At 24h post-transfection, harvest cell lysates to quantify Spike protein expression by Western blot or ELISA.
- Co-culture transfected APCs with autologous or matched peripheral blood mononuclear cells (PBMCs) for 5-7 days.
- Assess T-cell activation by:
- Intracellular Cytokine Staining (ICS): Stimulate co-culture with PMA/ionomycin or peptide pools, block secretion, stain for surface markers (CD3, CD4, CD8) and intracellular cytokines (IFN-γ, TNF-α).
- ELISpot: Seed T-cells from co-culture onto anti-IFN-γ coated plates, re-stimulate with Spike peptides, and quantify spot-forming units (SFUs).
- Analyze by flow cytometry or ELISpot reader.
Diagram 2: mRNA-LNP Immune Activation Pathway (86 chars)
Case Study: CAR-T Cell Therapy for B-cell Malignancies
Therapeutic Agent: Tisagenlecleucel (Kymriah) – Autologous CD19-directed CAR-T cells. Delivery Vector: Lentiviral Vector (LV) – a viral vector. Principle: Patient T-cells are genetically modified ex vivo using a self-inactivating lentivirus to express a chimeric antigen receptor (CAR) that targets CD19. The engineered cells are expanded and infused back into the patient, where they recognize and eliminate CD19+ B-cell malignancies.
Key Clinical Data: Table 3: Efficacy and Safety of Tisagenlecleucel in Relapsed/Refractory B-cell ALL (ELIANA Trial, NCT02435849)
| Parameter | Result at 3 Months | Long-term Follow-up |
|---|---|---|
| Overall Remission Rate (ORR) | 81% (95% CI: 71-89) | - |
| Complete Remission (CR) / CR with Incomplete Hematologic Recovery (CRi) | 60% CR, 21% CRi | |
| Relapse-free Survival | 80% at 6 months, 59% at 12 months | Median duration of remission not reached |
| Cytokine Release Syndrome (CRS) Incidence | 77% (any grade), 48% (grade 3/4) | Managed with tocilizumab (anti-IL-6R) |
| Neurologic Events Incidence | 40% (any grade), 21% (grade 3/4) | Typically reversible |
Experimental Protocol (Key Ex Vivo CAR-T Cell Manufacturing & Potency Assay):
- Objective: Generate and test the cytotoxic potency of CAR-T cells against CD19+ tumor cells.
- Materials: Patient leukapheresis product, anti-CD3/28 activation beads, recombinant IL-2, lentiviral vector encoding anti-CD19 CAR, target tumor cell lines (e.g., NALM-6), luciferase-based cytotoxicity assay kit, flow cytometer with anti-CAR detection reagent.
- Procedure:
- T-cell Activation: Isolate PBMCs, activate T-cells with anti-CD3/28 beads in serum-free media supplemented with IL-2 (50-100 IU/mL).
- Transduction: At 24h post-activation, transduce T-cells with lentiviral vector at a defined multiplicity of infection (MOI, e.g., 5-10) via spinoculation.
- Expansion: Culture cells for 7-14 days, maintaining cell density and IL-2 concentration. Monitor CAR expression by flow cytometry.
- Cytotoxicity Assay (Real-time Cell Analysis or Luciferase):
- Seed CD19+ target cells expressing luciferase in a 96-well plate.
- Add CAR-T cells at varying effector-to-target (E:T) ratios (e.g., 40:1, 20:1, 10:1, 5:1).
- Co-culture for 24-48 hours.
- Add luciferase substrate and measure luminescence. Cytotoxicity % = [1 - (Luminescencesample / Luminescencetargets_alone)] x 100.
- Cytokine Release: Collect supernatant from co-cultures and quantify IFN-γ, IL-2, etc., by ELISA.
Diagram 3: CAR-T Cell Manufacturing & Mechanism (70 chars)
The Scientist's Toolkit: Key Reagents for CAR-T Cell Research
| Research Reagent | Function & Rationale |
|---|---|
| Retronectin / Recombinant Fibronectin Fragment | Enhances lentiviral or retroviral transduction efficiency by co-localizing viral particles and target T-cells. |
| Anti-CD3/CD28 Magnetic Beads | Provides a reproducible, serum-free method for primary human T-cell activation and expansion, mimicking physiological stimulation. |
| Recombinant Human IL-2 | Critical growth factor for promoting the survival and proliferation of activated and transduced T-cells during ex vivo culture. |
| Flow Cytometry Anti-CAR Detection Reagent | Typically a recombinant protein (e.g., CD19-Fc or anti-idiotype antibody) to detect and quantify surface CAR expression on transduced T-cells. |
| Luciferase-Expressing Target Cell Lines | Enable sensitive, quantitative, and high-throughput measurement of CAR-T cell cytotoxic potency in vitro without radioactivity. |
Overcoming Delivery Barriers: Troubleshooting Low Efficiency and Improving Transfection
Within the broader thesis of advancing gene delivery vector research, understanding the fundamental principles that govern cellular uptake and transgene expression is paramount. This guide provides a systematic, technical framework for diagnosing the multifaceted causes of low transfection efficiency, a critical bottleneck in both viral and non-viral gene delivery research.
Transfection efficiency is the cornerstone metric for evaluating any gene delivery vector. Whether employing viral vectors (e.g., lentivirus, AAV) or non-viral methods (e.g., lipofection, electroporation), suboptimal efficiency stalls downstream applications. Diagnosis requires a holistic approach that considers the vector, the target cell, and their interaction.
Key Quantitative Benchmarks
The following table summarizes typical efficiency ranges for common transfection methods under optimal conditions.
Table 1: Typical Transfection Efficiency Ranges by Method
| Transfection Method | Typical Efficiency Range (in susceptible cell lines) | Primary Delivery Mechanism |
|---|---|---|
| Cationic Liposomes | 70-95% in vitro (e.g., HEK293) | Endocytosis, membrane fusion |
| Polyethylenimine (PEI) | 60-90% in vitro (e.g., HeLa) | Proton-sponge endosomal escape |
| Calcium Phosphate | 30-50% in vitro (adherent cells) | Precipitation & endocytosis |
| Electroporation | 50-80% (varies widely by cell type) | Membrane electropermeabilization |
| Lentiviral Transduction | >90% (dividing cells, with polybrene) | Receptor-mediated entry & integration |
| AAV Transduction | 30-70% (highly serotype & cell-dependent) | Receptor-mediated entry |
Step-by-Step Diagnostic Framework
Step 1: Verify Nucleic Acid Quality & Quantity
Protocol 1.1: Assessment of Plasmid DNA Purity and Integrity
- Measure A260/A280 and A260/A230 ratios via spectrophotometry (e.g., Nanodrop). Acceptable ranges: A260/A280 ~1.8-2.0; A260/A230 >2.0.
- Run 1 µg of plasmid on a 1% agarose gel. A predominant supercoiled (lower band) conformation is ideal. Significant smearing or a dominant linear/relaxed band indicates degradation or nicking.
- For viral preparations, quantify genomic titer via qPCR (for lentivirus, target the psi-packaging signal) and assess purity via SDS-PAGE for capsid proteins.
Table 2: Spectrophotometric Indicators of Nucleic Acid Quality
| Metric | Ideal Value | Indication if Out of Range |
|---|---|---|
| A260/A280 | 1.8 - 2.0 | <1.8: Protein/phenol contamination. >2.0: Possible RNA contamination. |
| A260/A230 | 2.0 - 2.2 | <2.0: Contamination by salts, chaotropes, or carbohydrates. |
Step 2: Optimize Vector-to-Cell Ratio & Formulation
The critical parameter for non-viral methods is the N/P ratio (molar ratio of Nitrogen (in cationic polymer) to Phosphate (in DNA)). For viral methods, it is the Multiplicity of Infection (MOI).
Protocol 2.1: Determining Optimal N/P Ratio for Polyplexes
- Prepare a master mix of plasmid DNA (e.g., 2 µg in 100 µL serum-free medium).
- In separate tubes, dilute cationic polymer (e.g., linear PEI, 1 mg/mL stock) in 100 µL serum-free medium to achieve N/P ratios from 1 to 20.
- Add the polymer solution to the DNA solution, vortex immediately, and incubate 15-30 min at RT for complex formation.
- Transfer complexes to cells. Include a fluorescent reporter plasmid (e.g., GFP) for easy quantification via flow cytometry 24-48 hours post-transfection.
- Plot transfection efficiency (%) vs. N/P ratio and cell viability (%) vs. N/P ratio to identify the optimal balance.
Protocol 2.2: Titrating Viral MOI
- Transduce target cells in the presence of a transduction enhancer (e.g., 8 µg/mL polybrene for lentivirus) with a serial dilution of viral supernatant.
- For vectors with a fluorescent marker, analyze by flow cytometry after 48-72 hours. For non-fluorescent vectors, use a functional assay (e.g., luciferase, antibiotic selection).
- Calculate MOI using the formula:
MOI = (Number of Transducing Units per mL * Volume in mL) / Number of Cells. Aim for the lowest MOI that gives desired efficiency to minimize off-target effects.
Step 3: Assess Cellular Health and State
Cell passage number, confluency, and metabolic state dramatically impact efficiency.
Protocol 3.1: Standardized Cell Preparation for Transfection
- Culture cells in recommended medium with serum. Avoid antibiotics during transfection.
- For adherent cells: Passage at 70-90% confluency. Seed for transfection at 50-70% confluency 18-24 hours prior. Using cells beyond passage 30 increases variability.
- For suspension cells: Maintain in logarithmic growth phase. Adjust seeding density to ensure optimal growth during the experiment.
- Viability Check: Mix 10 µL cell suspension with 10 µL Trypan Blue. Count live (unstained) and dead (blue) cells on a hemocytometer. Viability should be >95% pre-transfection.
Step 4: Investigate Intracellular Barriers
Successful delivery requires overcoming intracellular hurdles: endosomal entrapment, cytosolic degradation, and (for non-integrating vectors) nuclear import.
Diagram: Major Intracellular Barriers to Gene Delivery
Protocol 4.1: Assessing Endosomal Escape Using a Gal8-mCherry Assay
- Seed cells on imaging dishes to reach 50% confluency.
- Co-transfect cells with your gene delivery formulation and a plasmid encoding Galactosyltransferase 8 (Gal8) fused to mCherry. Gal8 binds exposed β-galactosyl sugars on disrupted endosomes.
- At 4-6 hours post-transfection, fix cells and image via confocal microscopy.
- Analysis: Co-localization of the fluorescent vector signal with bright Gal8-mCherry puncta indicates endosomal entrapment. Successful escape is indicated by diffuse cytosolic vector signal without Gal8 co-localization.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Transfection Optimization & Diagnosis
| Reagent/Category | Example Product/Brand | Primary Function in Diagnosis |
|---|---|---|
| High-Purity DNA Prep Kits | EndoFree Plasmid Kits (Qiagen), ZymoPure (Zymo Research) | Eliminate endotoxins that trigger cellular responses and reduce efficiency. |
| Fluorescent Reporter Plasmids | pEGFP-N1, pmCherry-C1, pGL4 Luciferase | Visual and quantitative assessment of transfection success and kinetics. |
| Cationic Liposome/Polymer | Lipofectamine 3000 (Thermo), jetOPTIMUS (Polyplus), linear PEI (Polysciences) | Gold-standard positive controls for non-viral delivery in optimization experiments. |
| Transduction Enhancers | Polybrene (Hexadimethrine Bromide), Vectofusin-1 (Miltenyi) | Increase viral vector attachment to cell surface, standardizing and boosting transduction. |
| Viability/Cytotoxicity Assays | MTT, CellTiter-Glo (Promega), LDH Release Assays | Quantify cellular health post-transfection to differentiate toxicity from poor delivery. |
| Endosomal Escape Assay | Gal8-mCherry plasmid (Addgene) | Visualize endosomal membrane damage as a proxy for successful escape. |
| Nuclear Staining Dyes | Hoechst 33342, DAPI | Delineate nuclei to assess nuclear import of vectors (via microscopy). |
| qPCR Reagents for Titering | SYBR Green or TaqMan assays targeting vector genomes | Accurately quantify physical and functional titers of viral vector preps. |
Step 5: Validate with a Controlled Experimental Workflow
A systematic workflow isolates variables and pinpoints failure points.
Diagram: Diagnostic Workflow for Low Transfection
Diagnosing low transfection efficiency is a rigorous exercise in applying first principles of gene delivery. By sequentially interrogating the quality of the genetic cargo, the stoichiometry of complex formation, the health of the target cell, and the intracellular fate of the vector, researchers can transform a vague experimental failure into a precise, solvable problem. This systematic approach directly informs the core thesis of vector research: that iterative, mechanistic problem-solving is essential for evolving the next generation of efficient and specific gene delivery vehicles.
Within the broader thesis on the basic principles of viral and non-viral gene delivery vectors, optimizing the production and quality control of viral vectors remains a cornerstone for successful gene therapy and vaccine development. This guide provides an in-depth technical analysis of strategies to enhance vector titers and infectivity, which are critical for efficacy, safety, and commercial viability.
Upstream Production Optimization
The goal of upstream processes is to maximize the yield of functional vector particles per volume of culture.
1.1. Cell Line and Culture Parameters Selecting the appropriate producer cell line (e.g., HEK293, HEK293T, Sf9) is paramount. Adherent vs. suspension culture significantly impacts scalability. Recent trends favor suspension-adapted HEK293 lines in serum-free media for high-density bioreactor production.
1.2. Transfection and Infection Strategies For transient transfection (common for lentivirus and AAV), the following are critical:
- DNA Quality: Use endotoxin-free plasmid preparation kits.
- Transfection Reagent: Optimize the polyethylenimine (PEI) to DNA ratio. New polymer-based reagents offer improved efficiency.
- Timing: Harvest timepoints must be optimized for each vector system to balance titer and particle integrity.
For baculovirus/Sf9 systems (common for large-scale AAV), optimizing the Multiplicity of Infection (MOI) and time of harvest is essential.
Table 1: Impact of Culture Parameters on Viral Titer
| Parameter | Low/Suboptimal Condition | High/Optimal Condition | Typical Effect on Functional Titer (Log Scale) |
|---|---|---|---|
| Cell Viability at Harvest | <80% | >90% | -1.0 to -2.0 |
| Dissolved Oxygen | <20% saturation | 40-60% saturation | -0.5 to -1.5 |
| pH Fluctuation | > ±0.5 units | Maintained at 7.2 ± 0.2 | -0.5 to -1.0 |
| Transfection Efficiency | <70% | >90% | -1.0 to -2.0 |
| Harvest Timepoint | Too Early/Late | Vector-specific optimum | -1.5 to -3.0 |
Protocol 1.1: High-Efficiency PEI-Mediated Transfection for LV Production in Suspension HEK293
- Day 0: Seed HEK293 suspension cells at 0.5–1.0 x 10^6 cells/mL in serum-free medium in a shake flask.
- Day 1: At cell density of 2.0–3.0 x 10^6 cells/mL, prepare transfection mix.
- A. DNA Mix: Dilute transfer, packaging, and envelope plasmids (typical ratio 1:1:1, mass optimized) in 1/10th of the final culture volume of fresh medium.
- B. PEI Mix: Dilute linear PEI (1 mg/mL stock, pH 7.0) in an equal volume of fresh medium as the DNA mix. Use a DNA:PEI ratio of 1:3 (w/w) as a starting point.
- Rapidly mix the PEI solution into the DNA solution. Vortex briefly and incubate for 10-15 minutes at room temperature.
- Add the DNA-PEI complexes dropwise to the cell culture with gentle swirling.
- Incubate at 37°C, 5% CO2, with shaking. Optionally, add enhancers like valproic acid or sodium butyrate 6-12 hours post-transfection.
- Harvest supernatant containing lentiviral vectors at 48-72 hours post-transfection.
Downstream Purification and Concentration
Purification removes empty capsids, cellular debris, and process-related impurities, directly impacting infectivity.
2.1. Chromatography Methods
- Affinity Chromatography: The gold standard for AAV purification using AVB Sepharose or POROS CaptureSelect resins that bind to intact capsids.
- Ion-Exchange Chromatography (IEX): Useful for polishing and separating full vs. empty capsids based on surface charge differences.
- Size-Exclusion Chromatography (SEC): Final polishing step to remove aggregates and exchange buffer.
2.2. Tangential Flow Filtration (TFF) Used for concentration and buffer exchange. Membrane molecular weight cutoff (MWCO) and cross-flow rate must be optimized to prevent vector shear and aggregation.
Quality Control and Potency Assessment
Rigorous QC is non-negotiable for clinical-grade vectors. Key metrics are summarized below.
Table 2: Essential Quality Control Assays for Viral Vectors
| Assay Category | Specific Assay | Target Metric | Acceptable Range (Example) | Method |
|---|---|---|---|---|
| Physical Titer | qPCR/ddPCR (genome copies) | Total Vector Genomes (vg/mL) | Varies by application | ISO/ICH compliant digital PCR |
| ELISA (capsid) | Total Capsids (cp/mL) | Varies by application | Capsid-specific antibody | |
| Infectivity/Potency | Transduction Assay (Flow Cytometry) | Transducing Units (TU/mL) | TU:vg ratio >1:100 (LV) | Reporter gene expression |
| TCID50 / Plaque Assay | Infectious Units (IU/mL) | Varies by vector | Cell-based infectivity | |
| Purity & Safety | SDS-PAGE / Western Blot | Protein Purity | Minimal host cell protein | Coomassie, silver stain |
| HCP/HCD ELISA | Host Cell Protein/DNA | <100 ng/dose (HCP), <10 ng/dose (HCD) | ELISA, qPCR | |
| Endotoxin (LAL) | Endotoxin Units (EU/mL) | <5 EU/kg body weight | Limulus Amebocyte Lysate | |
| Characterization | Empty/Full Capsid Analysis | % Full Capsids | >70% (desired for AAV) | Analytical Ultracentrifugation, ELISA, AUC |
| Sequencing | Vector Genome Integrity | 100% match to design | Next-Generation Sequencing |
Protocol 3.1: Flow Cytometry-Based Transduction Assay for Lentivirus (TU/mL)
- Cell Preparation: Seed HEK293T or other permissive cells in a 24-well plate at 1 x 10^5 cells/well.
- Virus Dilution: Prepare serial log dilutions (e.g., 10^-3 to 10^-6) of the lentiviral vector stock in complete medium containing polybrene (8 µg/mL).
- Transduction: Aspirate medium from cells and add 0.5 mL of each virus dilution per well. Include a "no virus" control well.
- Incubation: Incubate for 72 hours at 37°C.
- Analysis: Harvest cells using trypsin, wash with PBS, and resuspend in flow cytometry buffer. Analyze the percentage of cells expressing the reporter (e.g., GFP) using a flow cytometer.
- Calculation: Calculate Transducing Units per mL (TU/mL) using the formula:
TU/mL = (F × C × D) / V. Where F is the fraction of GFP+ cells, C is the total number of target cells at transduction, D is the virus dilution factor, and V is the volume of inoculum (mL). Use data from the dilution where F is between 2% and 20% for linear accuracy.
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function & Rationale |
|---|---|
| Polyethylenimine (PEI), Linear, 25kDa | Cationic polymer for transient transfection; forms complexes with DNA, facilitating cellular uptake. High efficiency and cost-effective for large-scale production. |
| Endotoxin-Free Plasmid Kits | Isolate high-purity plasmid DNA with minimal endotoxin contamination, which can reduce transfection efficiency and induce immune responses in final products. |
| POROS CaptureSelect AAVX Affinity Resin | Affinity chromatography resin for one-step purification of multiple AAV serotypes. Binds intact capsids, dramatically improving full/empty ratio and purity. |
| Sybr Safe DNA Gel Stain | Safer, more sensitive alternative to ethidium bromide for visualizing DNA in gels during quality control of viral genomes or residual host cell DNA. |
| Recombinant Human Fibronectin Fragment (RetroNectin) | Enhances transduction efficiency of retroviral and lentiviral vectors by co-localizing vector particles and target cells, crucial for ex vivo gene therapy protocols. |
| Digital PCR (ddPCR) Master Mix | Provides absolute quantification of vector genome copies without a standard curve. Essential for accurate and reproducible physical titer determination. |
| LAL Endotoxin Assay Kit | Based on Limulus Amebocyte Lysate, detects bacterial endotoxins at picogram levels, a critical safety release test for clinical-grade vectors. |
| Anti-AAV Capsid ELISA Kit | Quantifies total AAV capsid protein concentration, allowing calculation of the particle-to-infectivity ratio and empty/full capsid assessment. |
Visualization: Production and QC Workflow
Viral Vector Production & QC Workflow
Optimizing viral vector titers and infectivity requires a holistic approach integrating upstream cell culture, transfection, downstream purification, and rigorous, multi-parametric quality control. The methodologies and data presented here, framed within the fundamental principles of gene delivery vector research, provide a roadmap for researchers and developers to enhance the yield, quality, and therapeutic potential of their viral vector batches. Continuous innovation in production platforms and analytical techniques remains vital for advancing the field of gene therapy.
Gene therapy and nucleic acid-based therapeutics represent a paradigm shift in modern medicine. The core challenge lies in the efficient, safe, and targeted delivery of genetic material (DNA, mRNA, siRNA) into specific cells. Delivery vectors are broadly categorized into viral and non-viral systems. Viral vectors (e.g., lentivirus, AAV) leverage evolved biological mechanisms for high transduction efficiency but face significant limitations regarding immunogenicity, insertional mutagenesis risk, payload capacity, and complex manufacturing. Non-viral vectors—encompassing lipid nanoparticles (LNPs), polymeric nanoparticles, peptide-based systems, and inorganic nanoparticles—offer advantages in safety, design flexibility, large-scale production, and larger payload capacity. However, their clinical translation has been hampered by inferior transfection efficiency, stemming primarily from poor stability in biological fluids and inefficient cellular uptake and endosomal escape. This whitepaper, framed within the foundational thesis of gene delivery vector principles, provides an in-depth technical guide to cutting-edge strategies for enhancing the stability and uptake of non-viral vectors.
Core Challenges: Stability and Uptake Barriers
Stability Challenges
Non-viral vectors encounter multiple stability barriers:
- Physicochemical Instability: Aggregation, fusion, or payload leakage during storage or circulation.
- Serum Instability: Opsonization by serum proteins, leading to rapid clearance by the mononuclear phagocyte system (MPS).
- Nuclease Degradation: Susceptibility of nucleic acid payloads to enzymatic degradation (e.g., serum nucleases).
Uptake & Intracellular Trafficking Barriers
The journey from administration to target site involves sequential hurdles:
- Cellular Binding: Limited association with target cell membranes.
- Internalization: Inefficient entry, often reliant on suboptimal endocytic pathways.
- Endosomal Entrapment & Lysosomal Degradation: Failure to escape endosomes leads to payload degradation.
- Cytoplasmic Transport & Nuclear Entry: Inefficient trafficking to the site of action (cytosol for mRNA/siRNA, nucleus for DNA).
Strategic Approaches & Experimental Data
Enhancing Serum and Colloidal Stability
Strategy 1: Surface PEGylation The conjugation of poly(ethylene glycol) (PEG) creates a hydrophilic steric barrier, reducing opsonization and MPS clearance.
Table 1: Impact of PEGylation on LNP Stability and Pharmacokinetics
| PEG Lipid Molar % | Particle Size (nm) ± SD | PDI ± SD | Serum Half-life (min) in Mice | Liver Accumulation (%ID/g, 1h) |
|---|---|---|---|---|
| 0% (Non-PEGylated) | 145 ± 12 | 0.22 ± 0.03 | <5 | 85 ± 7 |
| 1.5% | 152 ± 10 | 0.18 ± 0.02 | 45 ± 6 | 65 ± 5 |
| 3.0% | 155 ± 8 | 0.15 ± 0.02 | 120 ± 15 | 35 ± 4 |
| 5.0% | 160 ± 15 | 0.19 ± 0.03 | 95 ± 10 | 50 ± 6 |
Data synthesized from recent studies on siRNA-LNPs (2023-2024). %ID/g = Percentage of Injected Dose per gram of tissue.
Protocol: Synthesis and Characterization of PEGylated LNPs
- Materials: Ionizable lipid (e.g., DLin-MC3-DMA), phospholipid (DSPC), cholesterol, PEG-lipid (DMG-PEG2000), nucleic acid payload (siRNA/mRNA), ethanolic buffer (pH 4.0), PBS (pH 7.4).
- Microfluidic Formulation: Load lipid mix (in ethanol) and aqueous nucleic acid buffer (in citrate) into separate syringes. Utilize a staggered herringbone or Y-junction microfluidic chip. Set total flow rate (TFR) to 10-12 mL/min and flow rate ratio (FRR, aqueous:organic) of 3:1 for rapid mixing. Collect nanoparticles in a PBS receiving vial.
- Dialyzation & Characterization: Dialyze against PBS (pH 7.4) for 2 hours to remove ethanol and buffer exchange. Characterize particle size and polydispersity index (PDI) via dynamic light scattering (DLS), zeta potential via electrophoretic light scattering, and encapsulation efficiency using a Ribogreen assay.
Strategy 2: Zwitterionic Lipid Coatings Emerging as alternatives to PEG, zwitterionic materials (e.g., carboxybetaine, phosphorylcholine) impart a "super-hydrophilic" surface, demonstrating superior anti-fouling properties and reduced accelerated blood clearance (ABC) phenomenon associated with PEG.
Strategy 3: Stabilizing Excipients Incorporation of antioxidants (e.g., α-tocopherol) or cholesterol analogs can improve lipid bilayer integrity and prevent oxidation or fusion during storage.
Enhancing Cellular Uptake and Targeted Delivery
Strategy 1: Ligand Conjugation for Active Targeting Surface functionalization with targeting ligands (peptides, antibodies, aptamers, small molecules) enables receptor-mediated endocytosis into specific cell types.
Table 2: Efficacy of Targeted vs. Non-Targeted Polymeric Nanoparticles
| Nanoparticle Type | Targeting Ligand | Receptor | Cell Line | Uptake (RFU) ± SD | Gene Knockdown (% vs Control) ± SD |
|---|---|---|---|---|---|
| PEI-PLGA NP (Non-Targeted) | N/A | N/A | HeLa (EGFR+) | 1,250 ± 210 | 22 ± 5 |
| PEI-PLGA NP (Targeted) | GE11 Peptide | EGFR | HeLa (EGFR+) | 8,750 ± 950 | 78 ± 7 |
| PEI-PLGA NP (Targeted) | GE11 Peptide | EGFR | HEK293 (EGFR low) | 1,450 ± 300 | 25 ± 6 |
| PEI-PLGA NP (Targeted) | Folate | Folate Receptor | HeLa (EGFR+) | 2,100 ± 400 | 30 ± 8 |
RFU = Relative Fluorescence Units (quantified via flow cytometry). Data from recent polymeric nanoparticle studies (2024).
Protocol: Conjugation of Targeting Ligands via Click Chemistry
- Materials: DBCO-modified nanoparticles (synthesized with DBCO-PEG-lipid or DBCO-terminated polymer), Azide-functionalized ligand, PBS (pH 7.4).
- Procedure: Purify DBCO-nanoparticles via size exclusion chromatography. Incubate with a 5-10 molar excess of azide-ligand in PBS at room temperature for 24 hours under gentle agitation. The strain-promoted alkyne-azide cycloaddition (SPAAC) reaction proceeds without catalyst. Purify the ligand-conjugated nanoparticles via dialysis or SEC to remove unreacted ligand.
- Validation: Confirm conjugation via change in zeta potential, HPLC analysis of reaction supernatant, or specific cell uptake assays using a receptor-positive vs. receptor-negative cell line.
Strategy 2: Charge Modulation While positive surface charge (cationic lipids/polymers) enhances initial cell membrane interaction, it also increases nonspecific binding and toxicity. A balance is achieved using ionizable lipids (positively charged at low pH during formulation, neutral at physiological pH) or charge-reversal polymers.
Promoting Endosomal Escape
Endosomal escape is the most critical bottleneck. The "proton sponge" effect of polymers like polyethylenimine (PEI) is one mechanism, but newer strategies are more efficient.
Strategy 1: pH-Sensitive Ionizable Lipids The cornerstone of modern LNPs. These lipids are neutral at physiological pH but gain positive charge in the acidic endosome (pH ~5.5-6.5). This promotes interaction with anionic endosomal membranes, leading to bilayer destabilization and payload release.
Diagram 1: Ionizable Lipid-Mediated Endosomal Escape Pathway (85 characters)
Strategy 2: Fusogenic Peptides Incorporating peptides derived from viral fusion proteins (e.g., HA2 peptide from influenza hemagglutinin) can directly mediate membrane fusion.
Protocol: In Vitro Endosomal Escape Assay Using Split GFP
- Materials: HeLa cells, split GFP system (CellLight BacMam 2.0 or similar), non-viral vector, confocal microscope.
- Procedure: Seed cells in an imaging chamber. Transduce cells with a baculovirus encoding a GFP strand targeted to the endosome (e.g., GFP1-10). 24h later, transfect with nanoparticles co-encapsulating your nucleic acid payload and the complementary GFP strand (GFP11). Successful endosomal escape allows the two GFP fragments to reconstitute in the cytosol, producing fluorescence. Image live cells 24-48h post-transfection. Quantify cytosolic fluorescence intensity per cell.
Advanced Designs and Future Directions
Multi-Functional Vectors: Combining stability, targeting, and endosomal escape features in a single platform. Example: An LNP with a zwitterionic outer layer, a GE11 peptide for tumor targeting, and a pH-triggered, pore-forming peptide for escape.
Stimuli-Responsive Vectors: Beyond pH, vectors responsive to redox potential (GSH in cytosol), enzymes (matrix metalloproteinases in tumor microenvironments), or light are under active investigation.
Machine Learning-Guided Design: In silico screening of chemical libraries to predict novel ionizable lipid structures with optimal pKa, biodegradability, and efficacy, dramatically accelerating lead candidate identification.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Non-Viral Vector Research
| Reagent/Material | Supplier Examples | Key Function in Research |
|---|---|---|
| Ionizable Cationic Lipids | Avanti, BroadPharm, MedChemExp | Core component of modern LNPs; enables efficient encapsulation and endosomal escape. |
| DMG-PEG2000 / DSG-PEG2000 | Avanti, NOF America | PEG-lipid used for LNP stabilization and surface functionalization. |
| DOPE / DSPC | Avanti, Sigma-Aldrich | Helper phospholipids for structuring lipid bilayers and promoting fusogenicity. |
| Cholesterol | Avanti, Sigma-Aldrich | Stabilizes lipid bilayer, modulates fluidity, and enhances in vivo efficacy. |
| Branched PEI (25 kDa) | Polysciences, Sigma-Aldrich | Gold-standard cationic polymer for in vitro transfections; strong proton sponge effect. |
| Microfluidic Chips | Dolomite, Precision NanoSystems | For reproducible, scalable preparation of uniform nanoparticles (LNPs, polymeric NPs). |
| Ribogreen Assay Kit | Invitrogen | Fluorometric quantification of nucleic acid encapsulation efficiency. |
| CellLight BacMam Reagents | Invitrogen | For labeling cellular organelles (endosomes, lysosomes) to study intracellular trafficking. |
| HPLC Systems & Columns | Agilent, Waters | For purity analysis of lipids, polymers, and characterization of final nanoparticle formulations. |
The development of effective gene delivery vectors, both viral and non-viral, represents a cornerstone of modern therapeutic biotechnology. The core thesis of this field posits that the ideal vector must achieve a critical balance: maximizing the efficiency of nucleic acid transfer and transgene expression while minimizing adverse host reactions, namely cytotoxicity and immune activation. This whitepaper provides an in-depth technical analysis of the mechanisms underlying these safety concerns and outlines contemporary strategies and methodologies for their mitigation, thereby advancing the fundamental principles of vector design.
Mechanisms of Cytotoxicity and Immune Activation
Viral Vectors
Adeno-Associated Viruses (AAVs) and Lentiviruses (LVs) are widely used but elicit distinct responses. AAVs can trigger innate immune sensing via TLR9 and TLR2-MyD88 pathways upon capsid engagement, leading to IFN-γ and TNF-α production. Adaptive immune responses to capsid and transgene products can eliminate transduced cells. Lentiviruses, while integrating, may activate pathogen-associated molecular pattern (PAMP) sensors, and insertional mutagenesis remains a genotoxic risk.
Non-Viral Vectors
Cationic lipids and polymers, such as Lipofectamine and polyethylenimine (PEI), induce cytotoxicity primarily through:
- Membrane disruption: Positive charge interaction with anionic cellular membranes.
- Reactive Oxygen Species (ROS) generation: Leading to oxidative stress and apoptosis.
- Inflammasome activation: Particularly the NLRP3 inflammasome by particulate materials, resulting in IL-1β release.
Diagram Title: Pathways of Vector-Induced Immune and Cytotoxic Responses
Quantitative Comparison of Vector Safety Profiles
The following table summarizes key safety parameters for common vector classes, based on recent in vitro and preclinical data.
Table 1: Comparative Safety and Immunogenicity Profile of Gene Delivery Vectors
| Vector Class | Specific Type | Primary Cytotoxicity Mechanism | Key Immune Sensors | Typical Inflammatory Cytokine Elevation (in vitro) | Risk of Insertional Mutagenesis |
|---|---|---|---|---|---|
| Viral | Adenovirus (Ad5) | High viral protein expression | TLR9, Cytosolic DNA sensors | High (IL-6, TNF-α, IFN-γ) | Low |
| Viral | Adeno-Associated Virus (AAV8) | High-dose hepatotoxicity | TLR2, TLR9, Antibody-mediated | Moderate (IFN-γ) | Very Low |
| Viral | Lentivirus (VSV-G) | Low (depends on titer) | TLR7/8 (RNA), Antibody-mediated | Low-Moderate | Moderate |
| Non-Viral | Cationic Liposome (e.g., DLin-MC3-DMA) | Membrane disruption, ROS | NLRP3 Inflammasome | High (IL-1β, IL-6) | None |
| Non-Viral | Polyethylenimine (PEI, 25kDa) | Osmotic lysis, apoptosis | NLRP3, TLRs | Very High (pan-inflammatory) | None |
| Non-Viral | Lipid Nanoparticle (LNP) for mRNA | Membrane stress, ionizable lipid | NLRP3, IFN response | Moderate (IL-1β, IFN-α) | None |
Experimental Protocols for Assessing Safety
Protocol: ComprehensiveIn VitroCytotoxicity Screening
Objective: Quantitatively assess vector-induced cell death and metabolic impairment. Materials: HEK293T or primary target cells, vector preparation, control vehicle. Procedure:
- Cell Seeding: Seed cells in a 96-well plate at 5,000-10,000 cells/well and culture for 24h.
- Vector Treatment: Treat cells with a serial dilution of the vector (e.g., varying multiplicity of infection (MOI) or weight/volume of polymer). Include untreated and vehicle-only controls. Incubate for 24-72h.
- Viability Assay (MTT): Add MTT reagent (0.5 mg/mL final) to each well. Incubate for 4h at 37°C. Solubilize formed formazan crystals with DMSO. Measure absorbance at 570 nm.
- Membrane Integrity (LDH Release): Collect supernatant from treated cells. Mix with LDH assay reagent per manufacturer's protocol. Measure absorbance at 490 nm. Calculate % cytotoxicity relative to total lysis control.
- Apoptosis/Necrosis (Flow Cytometry): Harvest cells (including supernatant). Stain with Annexin V-FITC and Propidium Iodide (PI) in binding buffer. Analyze by flow cytometry within 1 hour (Annexin V+/PI-: early apoptosis; Annexin V+/PI+: late apoptosis/necrosis).
Diagram Title: In Vitro Cytotoxicity Screening Workflow
Protocol: Profiling Innate Immune Activation
Objective: Measure cytokine release and intracellular signaling pathway activation post-vector exposure. Materials: Human peripheral blood mononuclear cells (PBMCs) or reporter cell lines (e.g., THP-1), vector, ELISA kits, phospho-specific antibodies. Procedure:
- Cell Stimulation: Seed PBMCs or reporter cells. Treat with vector or positive controls (e.g., LPS for TLR4, CpG for TLR9). Incubate for 6-24h.
- Cytokine Quantification (Multiplex ELISA): Collect cell culture supernatant. Analyze using a multiplex Luminex or ELISA array for cytokines (e.g., IL-1β, IL-6, TNF-α, IFN-α, IFN-γ).
- Signaling Pathway Analysis (Western Blot): Lyse cells at earlier timepoints (30min-2h). Resolve proteins by SDS-PAGE, transfer to membrane, and probe with antibodies against phospho-NF-κB p65, phospho-IRF3, phospho-p38 MAPK, and corresponding total proteins.
- Reporter Assay: Use HEK293 cells stably transfected with reporter constructs (e.g., NF-κB or ISRE luciferase). Treat with vector and measure luciferase activity after 6-24h.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Safety Assessment Experiments
| Reagent / Kit Name | Vendor Examples | Primary Function in Experiment |
|---|---|---|
| CellTiter 96 AQueous One MTT Assay | Promega, Thermo Fisher | Measures metabolic activity as a proxy for cell viability. |
| CytoTox 96 Non-Radioactive Cytotoxicity Assay (LDH) | Promega | Quantifies lactate dehydrogenase release from damaged cells. |
| Annexin V-FITC / PI Apoptosis Detection Kit | BioLegend, BD Biosciences | Distinguishes between apoptotic and necrotic cell populations via flow cytometry. |
| Human Proinflammatory Panel 13-plex Luminex Assay | Bio-Techne, Thermo Fisher | Simultaneously quantifies 13 key cytokines (IL-1β, TNF-α, IL-6, etc.) from small sample volumes. |
| Phospho-NF-κB p65 (Ser536) Antibody | Cell Signaling Technology | Detects activation of the canonical NF-κB pathway by Western blot. |
| NLRP3 Inhibitor (MCC950) | Sigma-Aldrich, Tocris | Highly specific chemical inhibitor to confirm NLRP3 inflammasome involvement. |
| Endotoxin Removal Resin (e.g., EndoTrap) | Hyglos, Thermo Fisher | Removes contaminating LPS from vector preparations to isolate vector-specific effects. |
| TLR9 Inhibitor ODN 2088 | InvivoGen | Synthetic oligonucleotide that selectively inhibits TLR9 signaling. |
Strategic Mitigation and Balancing Act
Vector Engineering Strategies
- Viral Vectors: Capsid Engineering (directed evolution, rational design) of AAVs to evade pre-existing immunity and reduce tropism for antigen-presenting cells. Promoter Optimization (use of tissue-specific or synthetic promoters) to limit off-target transgene expression and immune exposure.
- Non-Viral Vectors: Biodegradable Polymers (e.g., Poly(β-amino esters)) that degrade into less toxic metabolites. Ionizable Lipids (e.g., in LNPs) designed for endosomal escape at low pH while maintaining neutral charge in circulation, reducing nonspecific interactions.
Pharmacological and Formulation Approaches
- Immunomodulation: Transient co-administration of corticosteroids (e.g., dexamethasone) or TOR inhibitors (e.g., sirolimus) to dampen adaptive immune responses against the vector or transgene.
- Surface Functionalization: PEGylation or incorporation of CD47 mimetic peptides ("don't eat me" signals) to reduce phagocytic clearance and innate immune recognition.
Diagram Title: Strategic Approaches to Balance Safety & Efficiency
The pursuit of the optimal gene delivery vector is fundamentally an exercise in balancing two opposing forces: the necessity for high delivery efficiency and the imperative for biological safety. A deep mechanistic understanding of vector-specific cytotoxicity and immunogenicity, coupled with rigorous standardized in vitro and in vivo safety profiling as outlined, provides the roadmap. Continued innovation in vector engineering, formulation science, and adjunctive immunomodulation will be critical to translate the immense promise of gene therapy into safe, effective, and durable clinical realities, fulfilling the core thesis of the field.
The development of viral and non-viral vectors for gene therapy represents a cornerstone of modern translational medicine. A core thesis in gene delivery research posits that the fundamental principles of vector design—efficacy, specificity, and safety—must be rigorously preserved and controlled during the transition from small-scale proof-of-concept to large-scale clinical-grade manufacturing. This transition, or scale-up, is not merely a volumetric increase but a multidimensional challenge involving process engineering, quality assurance, and regulatory compliance. This guide details the technical and procedural hurdles encountered when moving from research-grade (mg, ≤1L cultures) to clinical-grade (g to kg, ≥200L bioreactors) vector production.
The disparities between research and clinical production are quantified across several key parameters.
Table 1: Comparative Analysis of Research-Grade vs. Clinical-Grade Production
| Parameter | Research-Grade (Lab-Scale) | Clinical-Grade (GMP) | Primary Challenge |
|---|---|---|---|
| Scale/Volume | 0.1 - 1 L culture | 50 - 2000 L bioreactor | Mixing, oxygenation, shear stress. |
| Cell Culture | Adherent (flasks, stacks) | Suspension (bioreactor) | Process change requires full re-optimization and new cell lines. |
| Upstream Yield | Variable, 1e10 - 1e11 VP/mL* | Must be consistent, >1e11 VP/mL | Reproducibility and titer maintenance at scale. |
| Purification | Ultracentrifugation, lab-columns | Tangential Flow Filtration, Chromatography | Scalability, resolution, yield loss. |
| Final Formulation | Simple buffers, often unstable | Defined, stable formulation (e.g., with cryoprotectants) | Long-term stability for global distribution. |
| Quality Control | Purity, titer (physical). | Full safety, potency, identity testing (e.g., RCA, replication-competent virus). | Implementing rigorous, validated release assays. |
| Documentation | Lab notebook. | Full Device Master File / Chemistry, Manufacturing, and Controls (CMC) section. | Traceability, standard operating procedures (SOPs). |
| Cost per Dose | High (inefficient process). | Must be commercially viable. | Process intensification and yield optimization. |
*VP = Viral Particles (for viral vectors).
Table 2: Common Viral Vectors and Their Specific Scale-Up Challenges
| Vector Type | Research Production Method | Key Clinical-Grade Challenge | Typical Clinical Titer Target |
|---|---|---|---|
| AAV | Transfection of HEK293 cells. | Scaling plasmid transfection; host cell DNA/protein clearance. | >1e14 vector genomes (vg)/L |
| Lentivirus | Transfection of HEK293T cells. | Biosafety (containment of replication-competent lentivirus), stability. | >1e8 TU/mL* |
| Adenovirus | Infection of HEK293 cells. | Controlling replication-competent adenovirus (RCA), sheer sensitivity. | >1e12 VP/mL |
*TU = Transducing Units.
Detailed Methodologies for Critical Scale-Up Experiments
Protocol: Optimization of AAV Production in Suspension Cells
Objective: Transition from adherent HEK293 cell transfection to scalable suspension culture for AAV production. Background: Plasmid transfection scales poorly. This protocol uses a stable producer cell line in suspension.
- Cell Line Development: Generate a HEK293-derived suspension cell line containing the rep/cap genes and the recombinant AAV genome with flanking ITRs, either inducible or stable.
- Bioreactor Inoculation: Seed a benchtop bioreactor (e.g., 3L working volume) with cells at 0.5e6 cells/mL in chemically defined, serum-free medium.
- Process Control: Maintain temperature at 37°C, pH at 7.2 (controlled with CO2 and base), dissolved oxygen (DO) at 40% via sparging, and agitation at 100-150 rpm to prevent shear damage.
- Induction/Harvest: At a cell density of 4-5e6 cells/mL, induce vector production (e.g., with temperature shift or chemical inducer like doxycycline). Harvest cells and supernatant 48-72 hours post-induction.
- Clarification: Use depth filtration (0.5/0.2 µm) to remove cell debris.
- Analytics: Sample for cell viability (trypan blue), total cell density, and vector genome titer (ddPCR) to calculate volumetric yield (vg/L).
Protocol: Validation of Clearance of Process-Related Impurities
Objective: Demonstrate that the purification process effectively removes host cell DNA (HCD) and host cell protein (HCP). Background: A critical CMC requirement for regulatory filing.
- Spike and Purify: Spike a known quantity of purified, radio- or fluorescently-labeled HCD and HCP into a mock harvest material.
- Process Simulation: Subject the spiked material to the entire downstream process (e.g., affinity chromatography, ion-exchange, TFF).
- Sample Analysis: Collect samples from each purification step (load, flow-through, wash, elution pools).
- Quantitation: For HCD, use qPCR with probes for highly repetitive genomic elements (e.g., Alu sequences for human). For HCP, use ELISA with antibodies against a broad spectrum of host proteins.
- Log Reduction Value (LRV) Calculation: LRV = log10 (Initial Impurity Load / Final Impurity in purified product). Target LRV for HCD is typically >4 log10.
Visualizing Key Concepts and Workflows
Scale-Up Process Comparison: Lab vs. GMP
Interlinked Challenges in Gene Therapy Scale-Up
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents & Materials for Process Development
| Item | Function in Scale-Up Context |
|---|---|
| Chemically Defined, Serum-Free Medium | Essential for suspension culture; ensures consistency, reduces risk of adventitious agents, and simplifies downstream purification. |
| GMP-Grade Plasmids or Cell Banks | Starting materials must be sourced from GMP-compliant facilities for Master Cell Bank (MCB) or Working Cell Bank (WCB) generation. |
| Affinity Chromatography Resins (e.g., AVB Sepharose for AAV9, Lentilectin for LV) | Critical for capture step; provides high purity and scalability compared to research methods like ultracentrifugation. |
| Tangential Flow Filtration (TFF) Systems | For concentration and buffer exchange at large volumes; more scalable and controllable than dialysis. |
| Process Analytics: ddPCR/QPCR | For accurate, absolute quantification of vector genome titer without a standard curve, crucial for process monitoring. |
| Host Cell Protein (HCP) ELISA Kits | Quantifies a major process-related impurity; kit must be specific to the host cell line used (e.g., HEK293). |
| Replication-Competent Virus (RCV) Assay Components | Critical safety test; requires specific cell lines (e.g., C8166 for LV) and detection methods (ELISA, PCR). |
| Stability Study Chambers | Controlled temperature and humidity chambers (2-8°C, -70°C, accelerated) to establish product shelf-life. |
Head-to-Head Analysis: Validating and Choosing Between Viral and Non-Viral Platforms
Within the foundational thesis of gene delivery vector research, a systematic comparison of viral and non-viral systems is paramount. This whitepaper provides an in-depth technical analysis of four core metrics—Transduction Efficiency, Cargo Capacity, Durability, and Safety—framing them as the critical determinants for vector selection in therapeutic and research applications. The evolution of vector engineering focuses on optimizing these often competing parameters.
Core Metrics: Definitions and Measurement
Transduction Efficiency: The percentage of target cells that successfully express the delivered transgene. Measured via flow cytometry (e.g., % GFP+ cells) or luminescence assays. Cargo Capacity: The maximum size of genetic material (in kilobases, kb) a vector can package and deliver without structural compromise. Durability: The persistence of transgene expression, categorized as transient (days to weeks) or stable (long-term integration). Safety: A composite profile including immunogenicity, risk of insertional mutagenesis, and off-target effects.
Quantitative Comparative Analysis
Table 1: Comparative Metrics of Major Viral Vectors
| Vector | Typical Transduction Efficiency In Vitro (%) | Cargo Capacity (kb) | Durability (Expression) | Primary Safety Concerns |
|---|---|---|---|---|
| Adenovirus (AdV) | 70-95 | 7-8 (up to 36 in gutless) | Transient (weeks) | High immunogenicity, pre-existing immunity |
| Adeno-Associated Virus (AAV) | 40-90 (serotype-dependent) | < 5 | Long-term but episomal | Limited immunogenicity, capsid toxicity at high dose |
| Lentivirus (LV) | 60-90 | ~8-10 | Stable (integrating) | Low immunogenicity, risk of insertional mutagenesis |
| Retrovirus (γ-RV) | 50-80 | ~8 | Stable (integrating) | High risk of insertional mutagenesis |
Table 2: Comparative Metrics of Non-Viral Vectors
| Vector / Method | Typical Transduction Efficiency In Vitro (%) | Cargo Capacity | Durability (Expression) | Primary Safety Concerns |
|---|---|---|---|---|
| Lipid Nanoparticles (LNPs) | 50-90 (mRNA), 10-70 (pDNA) | Virtually unlimited | Transient (days-weeks) | Reactogenicity, dose-dependent cytotoxicity |
| Polyethyleneimine (PEI) | 20-60 | High (>10 kb) | Transient | High cytotoxicity, aggregation in serum |
| Electroporation | 70-90 | High | Transient to stable (if CRISPR) | High cell mortality, requires ex vivo setting |
| Naked DNA / Physical | < 1 | High | Transient | Very low immunogenicity, extremely inefficient |
Experimental Protocols for Key Metrics
Protocol: Measuring Transduction Efficiency via Flow Cytometry
Objective: Quantify the percentage of cells expressing a delivered reporter gene (e.g., GFP). Materials: Transduced cell culture, flow cytometry buffer (PBS + 2% FBS), fixative (4% PFA, optional), flow cytometer. Procedure:
- Seed & Transduce: Seed target cells (e.g., HEK293, HeLa) in a 12-well plate. At 70% confluency, transduce with vector carrying GFP at a predefined MOI (Multiplicity of Infection). Include untransduced controls.
- Incubate: Incubate for 48-72 hours to allow for gene expression.
- Harvest: Trypsinize cells, quench with complete media, and pellet at 300 x g for 5 min.
- Wash & Resuspend: Wash cells once with flow buffer and resuspend in 500 µL of flow buffer. Optionally fix with 4% PFA for 15 min on ice if biosafety requires.
- Acquire Data: Analyze minimum 10,000 events per sample on a flow cytometer. Gate on live cells using forward/side scatter.
- Analyze: Determine the percentage of cells in the GFP-positive channel compared to the untransduced control.
Protocol: Assessing Cargo Capacity via Restriction Analysis
Objective: Confirm the integrity and size of packaged genetic cargo. Materials: Purified vector particles, lysis buffer (e.g., containing SDS/proteinase K), phenol-chloroform, ethanol, restriction enzymes, agarose gel electrophoresis system. Procedure:
- Extract Vector Genome: Incubate 50 µL of purified vector stock with lysis buffer for 1h at 56°C. Extract DNA using phenol-chloroform and precipitate with ethanol.
- Digest: Resuspend DNA. Set up restriction digests using enzymes that release the insert from the vector backbone.
- Electrophorese: Run digested and undigested samples on a 0.8% agarose gel alongside a high-molecular-weight DNA ladder.
- Analyze: Compare the size of the released insert band to the expected size. Smearing or bands smaller than expected indicate packaging failure or truncation.
Visualizing Key Pathways and Workflows
Title: Workflow for Measuring Transduction Efficiency
Title: Lentiviral Vector Cell Entry and Integration Pathway
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Vector Characterization Experiments
| Reagent / Kit | Primary Function | Example Vendor(s) |
|---|---|---|
| Lenti-X Concentrator | Rapid concentration of lentiviral supernatants via precipitation. | Takara Bio |
| AAVpro Purification Kit | Purification of AAV serotypes using affinity chromatography. | Takara Bio |
| Lipofectamine 3000 | A leading lipid-based transfection reagent for plasmid DNA delivery. | Thermo Fisher |
| CellTiter-Glo Luminescent Assay | Quantifies cell viability/metabolism post-transduction. | Promega |
| QuickTiter Lentivirus Assay Kit | Quantifies lentiviral particles (p24) and infectious units. | Cell Biolabs |
| DNase I (RNase-free) | Treatment of vector prep to remove un-packaged DNA prior to genome titering. | NEB, Roche |
| CRISPR-Cas9 RNP Complex | For non-viral gene editing; Cas9 protein + gRNA for electroporation. | IDT, Synthego |
| Polybrene (Hexadimethrine bromide) | Cationic polymer that enhances viral transduction by neutralizing charge repulsion. | Sigma-Aldrich |
| Puromycin Dihydrochloride | Selection antibiotic for stable cell line generation post-integration. | Thermo Fisher |
| Human IFN-γ ELISpot Kit | Measures T-cell immunogenicity against viral capsids. | Mabtech |
Within the paradigm of gene delivery vector research, validation assays form the critical bridge between vector design and therapeutic application. This guide details the core quantitative assays required to establish the efficacy of both viral and non-viral vectors, framed by the central thesis that rigorous, multi-layered validation is a fundamental principle for advancing the field from benchtop to bedside.
Quantifying Delivery: Physical Uptake and Intracellular Fate
The first validation tier measures the vector's ability to reach and enter target cells.
Key Assays & Data
Table 1: Assays for Quantifying Vector Delivery
| Assay | Principle | Quantitative Readout | Key Considerations |
|---|---|---|---|
| qPCR/ddPCR for Vector Genomes | DNA isolation followed by amplification of vector-specific sequences. | Vector genomes per cell (VG/cell) or per µg DNA. | Distinguishes internalized from membrane-bound vector; does not indicate functionality. |
| Flow Cytometry (Labeled Vector) | Direct detection of fluorophore-conjugated vectors (e.g., Cy5-labeled LNPs) or immunostaining of capsid proteins. | Percentage of positive cells; mean fluorescence intensity (MFI). | Measures cellular association/uptake; fluorophore conjugation may alter vector properties. |
| Biodistribution Imaging | In vivo tracking using bioluminescence (luciferase), radiolabels (e.g., ⁹⁹ᵐTc, ¹²⁵I), or NIR fluorophores. | Signal intensity per organ (Radiance [p/s/cm²/sr] or %ID/g). | Critical for in vivo studies; correlates physical delivery to target vs. off-target tissues. |
Detailed Protocol: Determination of Vector Genomes per Cell via ddPCR
- Materials: Cell lysate or purified genomic DNA, ddPCR Supermix for Probes (no dUTP), primer/probe set targeting a vector-specific sequence (e.g., WPRE, polyA signal), nuclease-free water, droplet generator, and a QX200/QX600 droplet reader.
- Method:
- Sample Prep: Harvest transduced cells (e.g., 72h post-transduction). Isolate total genomic DNA using a silica-membrane column kit. Accurately quantify DNA concentration.
- Reaction Setup: Prepare a 20µL reaction mix per sample: 10µL 2x ddPCR Supermix, 1µL of each primer (900nM final), 0.25µL of probe (250nM final), ~50ng of sample DNA, and nuclease-free water to volume.
- Droplet Generation: Transfer 20µL of the reaction mix and 70µL of Droplet Generation Oil for Probes into the DG8 cartridge. Generate droplets using the QX200 Droplet Generator.
- PCR Amplification: Transfer 40µL of emulsified droplets to a 96-well PCR plate. Seal and run PCR: 95°C for 10 min (enzyme activation), then 40 cycles of 94°C for 30s and 60°C for 1 min (annealing/extension), with a ramp rate of 2°C/s. A final 98°C step for 10 min enzyme deactivation.
- Droplet Reading & Analysis: Load plate into the droplet reader. Analyze using QuantaSoft software. The system counts positive (fluorescent) and negative droplets. Calculate vector genome copies/µL using Poisson statistics. Normalize to the mass of input DNA and the diploid genome equivalent of the target cell to derive VG/cell.
Quantifying Expression: Transgene Output
Successful delivery must lead to transgene expression, measured at the RNA and protein levels.
Key Assays & Data
Table 2: Assays for Quantifying Transgene Expression
| Assay | Target | Quantitative Readout | Key Considerations |
|---|---|---|---|
| RT-qPCR | Transgene-specific mRNA | Relative expression (2^(-ΔΔCt)) or absolute copy number via standard curve. | Requires RNA isolation and reverse transcription; sensitive to primer design; measures only transcript levels. |
| Flow Cytometry (Reporter Protein) | Detection of fluorescent (e.g., GFP) or cell surface (e.g., CD4) reporter proteins. | % Positive cells, MFI, geometric mean. | Single-cell resolution; excellent for heterogeneous populations; requires intracellular antigen for non-secreted proteins. |
| ELISA / Luminescence Assay | Secreted (e.g., hFIX) or intracellular enzymes (e.g., Luciferase). | Concentration (ng/mL) or Relative Light Units (RLU). | High throughput; sensitive; luminescence offers a broad dynamic range. |
Detailed Protocol: Flow Cytometry for Intracellular Reporter Expression
- Materials: Transduced cells, PBS, 4% paraformaldehyde (PFA), permeabilization buffer (e.g., 0.1% Triton X-100 in PBS), blocking buffer (e.g., 5% BSA in PBS), primary antibody specific to transgene protein, fluorophore-conjugated secondary antibody, flow cytometry staining buffer.
- Method:
- Harvest & Fix: Harvest cells (e.g., 96h post-transduction), wash with PBS, and resuspend in 4% PFA. Incubate for 15 min at room temperature (RT).
- Permeabilize & Block: Pellet cells, wash with PBS, and resuspend in ice-cold permeabilization buffer for 15 min. Wash cells and resuspend in blocking buffer for 30 min at RT.
- Stain: Pellet cells and resuspend in primary antibody diluted in staining buffer. Incubate for 1h at RT or overnight at 4°C. Wash cells twice with staining buffer.
- Secondary Stain: Resuspend cell pellet in fluorophore-conjugated secondary antibody (e.g., Alexa Fluor 488) diluted in staining buffer. Incubate for 45 min at RT in the dark. Wash twice.
- Acquisition & Analysis: Resuspend cells in PBS with 1% BSA. Acquire data on a flow cytometer (collect ≥10,000 events per sample). Analyze using FlowJo or similar: gate on live, single cells, then determine the percentage of positively staining cells and the MFI of the population.
Quantifying Functional Outcomes: Phenotypic Correction
The ultimate validation is the restoration of cellular or organismal function.
Key Assays & Data
Table 3: Assays for Quantifying Functional Outcomes
| Functional Context | Example Assay | Quantitative Readout |
|---|---|---|
| Gene Knockdown (siRNA/shRNA) | RT-qPCR of target mRNA; Western blot of target protein. | % Reduction in transcript or protein level vs. non-targeting control. |
| Gene Addition (Therapeutic Protein) | ELISA for secreted protein; Enzyme activity assay (e.g., β-glucuronidase). | Functional protein units (e.g., IU/mL) or enzymatic activity (nmol/min/mg). |
| Gene Editing (CRISPR-Cas) | T7 Endonuclease I or ICE/Synthego Analysis; NGS for indel frequency. | % Indel modification; % homology-directed repair (HDR). |
| Phenotypic Rescue (Disease Model) | In vitro rescue assay (e.g., chloride efflux for CFTR); In vivo survival or behavioral study. | Normalization of functional metric (e.g., efflux rate); Kaplan-Meier survival curve; improved behavioral score. |
Detailed Protocol: T7 Endonuclease I Assay for CRISPR Editing Efficiency
- Materials: Genomic DNA from edited cells, PCR primers flanking the target site, high-fidelity PCR master mix, T7 Endonuclease I enzyme, NEBuffer 2.1, agarose gel electrophoresis system.
- Method:
- PCR Amplification: Amplify the target genomic region (amplicon size ~500-800bp) using high-fidelity polymerase. Purify the PCR product.
- Heteroduplex Formation: Dilute purified PCR product to ~50ng/µL. Denature and reanneal in a thermocycler: 95°C for 5 min, ramp down to 85°C at -2°C/s, then ramp to 25°C at -0.1°C/s. This allows formation of heteroduplexes from mismatched wild-type and edited strands.
- Digestion: Prepare a 20µL digestion reaction: 200ng of reannealed PCR product, 2µL NEBuffer 2.1, 0.5µL T7 Endonuclease I (or appropriate unit), and water. Incubate at 37°C for 30 min.
- Analysis: Run the digested product on a 2% agarose gel. Cleavage of heteroduplexes yields two smaller fragments. Quantify band intensities using ImageJ software. Calculate the indel frequency using the formula: % Indel = 100 × (1 - √(1 - (b + c)/(a + b + c))), where a is the integrated intensity of the undigested band, and b & c are the intensities of the cleavage products.
The Scientist's Toolkit: Essential Reagent Solutions
Table 4: Key Research Reagent Solutions for Validation Assays
| Reagent Category | Example Product/Kit | Primary Function in Validation |
|---|---|---|
| Nucleic Acid Quantification | Qubit dsDNA HS Assay Kit; Quant-iT RiboGreen RNA Assay | Accurate, selective quantification of DNA or RNA for normalization in qPCR/ddPCR and RT-qPCR. |
| Droplet Digital PCR | Bio-Rad ddPCR EvaGreen Supermix; PrimePCR ddPCR Assays | Absolute quantification of vector copy number or transcript copies with high precision, without a standard curve. |
| Flow Cytometry Antibodies | Anti-AAV Capsid Antibody (clone B1); Anti-GFP Alexa Fluor 488 Conjugate | Detection of viral particle uptake (primary) or intracellular transgene expression (secondary-conjugated). |
| Secreted Protein Detection | Human FIX/Factor IX Quantikine ELISA Kit; Renilla-Glo Luciferase Assay System | Sensitive, quantitative measurement of therapeutic protein secretion or enzymatic reporter activity. |
| Genome Editing Analysis | GeneArt Genomic Cleavage Detection Kit; Alt-R HDR Enhancer V2 | Detection of nuclease-induced indels (T7E1) or improvement of homology-directed repair rates. |
Visualizing Validation Workflows and Pathways
Diagram 1: Multi-Tier Gene Therapy Validation Cascade
Diagram 2: ddPCR Workflow for Vector Genome Quantification
Diagram 3: Functional Validation Pathway for Gene Editing
This technical guide analyzes the fundamental trade-offs in planning gene delivery vector projects, from basic research through clinical development. Framed within the core principles of viral and non-viral vector research, we provide a data-driven framework for strategic decision-making, emphasizing timelines, costs, and technical milestones.
The journey from vector concept to therapeutic involves a discontinuous shift in objectives. Research phases prioritize mechanistic understanding and in vitro efficacy, while clinical development demands stringent safety, scalable manufacturing, and regulatory compliance. This pivot dictates vastly different resource allocation and success metrics.
Quantitative Analysis: Research vs. Development
Table 1: Comparative Cost and Timeline Estimates (2023-2024 Data)
| Phase/Component | Typical Duration | Estimated Cost Range | Primary Objectives | Success Rate (Industry Avg.) |
|---|---|---|---|---|
| Basic Research (Vector Design & In Vitro) | 1-3 years | $250,000 - $1.5M | Proof-of-concept, mechanism, initial cytotoxicity | N/A (Feasibility-driven) |
| Preclinical Development | 1-2 years | $1M - $5M | In vivo efficacy (animal models), safety/toxicology (IND-enabling) | ~30% (From research lead to IND) |
| Phase I Clinical Trial | 1-2 years | $4M - $10M | Safety, tolerability, dosing in humans | ~50-60% (From IND to Phase II) |
| Phase II Clinical Trial | 2-3 years | $12M - $30M | Efficacy, side effect profiling | ~30% (From Phase I to Phase III) |
| Phase III Clinical Trial & Approval | 3-5 years | $50M - $150M+ | Confirmatory efficacy, large-scale safety, regulatory submission | ~60-70% (From Phase II to NDA/BLA) |
Table 2: Key Attribute Comparison: Viral vs. Non-Viral Vectors
| Attribute | Viral Vectors (e.g., AAV, Lentivirus) | Non-Viral Vectors (e.g., LNPs, Polymers) |
|---|---|---|
| Typical Transfection Efficiency | High (>80% in vitro) | Moderate to High (50-90%, formulation-dependent) |
| Payload Capacity | Limited (AAV: ~4.7kb; Lentivirus: ~8kb) | Large (>10kb) |
| Immunogenicity Risk | High (Pre-existing & induced immunity) | Generally Lower |
| Manufacturing Complexity (Scale-up) | High (Cell culture, purification, titering) | Moderate (Chemical synthesis, formulation) |
| Timeline to GMP-grade Material | 18-36 months | 12-24 months |
| Dominant Cost Driver (Preclinical) | Vector Production & Purification | Payload Synthesis & Formulation Optimization |
Experimental Protocols for Critical Milestones
Protocol 1:In VitroTransfection Efficiency & Cytotoxicity Benchmarking
Purpose: Standardized comparison of novel vector candidates against benchmarks. Materials: HEK293T or relevant primary cells, vector preparations, serum-free media, assay kits. Procedure:
- Seed cells in 96-well plates at 5,000 cells/well. Incubate for 24h.
- Prepare vector dilutions in serum-free medium (e.g., 0.1, 1, 10, 100 MOI or µg DNA/mL).
- Transfect using appropriate method (e.g., direct addition for viral vectors, lipofection mix for plasmids).
- At 48h post-transfection, harvest supernatant for cytotoxicity (LDH assay) and lyse cells for efficacy (luciferase/GFP quantification via plate reader).
- Normalize efficacy data to cell viability. Calculate IC50 and Transfection EC50.
Protocol 2:In VivoBiodistribution & Persistence (Mouse Model)
Purpose: IND-enabling assessment of vector trafficking and durability. Materials: C57BL/6 mice, vector (IV or local administration), IVIS imaging system, qPCR reagents. Procedure:
- Administer vector encoding a luciferase reporter via intended route (e.g., tail vein injection).
- At time points (Day 1, 7, 28, 84), perform in vivo bioluminescence imaging (IVIS) under anesthesia.
- Euthanize animals (n=3 per time point). Harvest organs (liver, spleen, lung, heart, target tissue).
- Isolate genomic DNA from homogenized tissues. Perform TaqMan qPCR for vector-specific sequence (e.g., polyA signal) and a host gene (e.g., mGapdh) for normalization.
- Report vector genome copies per µg host DNA. Correlate with IVIS signals.
Visualizing Key Pathways and Workflows
Diagram 1: Gene Delivery Vector R&D Decision Pathway (100 chars)
Diagram 2: AAV Vector Intracellular Trafficking Pathway (100 chars)
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Gene Delivery Vector Research
| Reagent/Material | Supplier Examples | Primary Function | Key Consideration |
|---|---|---|---|
| Polyethylenimine (PEI), linear | Polysciences, Sigma | Polymeric transfection reagent; condenses DNA/RNA via charge interaction. | Branching & molecular weight critically impact efficiency & toxicity. |
| Lipofectamine 3000 | Thermo Fisher | Cationic lipid-based transfection kit for in vitro DNA/RNA delivery. | Standard for non-viral efficiency benchmarking; optimized for many cell lines. |
| AD-Binder/HEK293 Transfection Kit | Cell Biolabs, Sirion Biotech | Production of high-titer recombinant AAV or lentivirus. | Essential for research-scale viral vector prep; includes transfection & purification resins. |
| DNase I, RNase-free | Roche, NEB | Degrades unpackaged nucleic acids during viral vector purification. | Critical for accurate titer determination (genomic vs. total particles). |
| LIVE/DEAD Viability/Cytotoxicity Kit | Thermo Fisher | Dual-fluorescence assay for simultaneous viability & cytotoxicity measurement. | Superior to MTT for transient transfection readouts. |
| pAAV-MCS (Multiple Cloning Site) Vector | Addgene | Standard plasmid backbone for AAV genome construction. | Contains ITRs essential for packaging; choice of promoter dictates tropism. |
| QuickTiter AAV Quantitation Kit | Cell Biolabs | ELISA-based detection of intact AAV particles (anti-capsid). | Measures functional titer independent of genome integrity. |
| Luciferase Assay System | Promega | Quantitative measurement of transfection/transduction efficacy. | Standard reporter; linear range >7 orders of magnitude. |
Within the broader thesis on the basic principles of viral and non-viral gene delivery vectors, navigating the regulatory landscape is a critical translational step. This guide details the approval pathways for major vector classes, integrating current regulatory frameworks, quantitative data, and experimental methodologies essential for preclinical development.
Regulatory Agencies & Primary Pathways
The core regulatory pathways for gene therapy products (GTPs) are defined by major agencies. The specific route depends on the product's nature, target indication, and clinical context.
Table 1: Primary Regulatory Pathways for Advanced Therapy Medicinal Products (ATMPs)
| Agency | Pathway | Key Criteria/Description | Typical Timeline (Calendar Days) |
|---|---|---|---|
| U.S. FDA | Biologics License Application (BLA) | Full approval for commercial marketing. Requires substantial evidence of safety & efficacy from adequate and well-controlled studies. | Standard: 180-360 (Priority Review: 180) |
| U.S. FDA | Investigational New Drug (IND) | Application to initiate clinical trials in humans. Must demonstrate sufficient preclinical pharmacology, toxicology, and CMC data. | FDA has 30 days to review for safety hold. |
| EMA (EU) | Marketing Authorization Application (MAA) | Centralized procedure approval for EU market. Comparable to FDA BLA. | Standard: 210 (Accelerated: 150) |
| EMA (EU) | Clinical Trial Application (CTA) | Submission to national Competent Authority and Ethics Committee to begin clinical trials. | ~60-90 days (varies by member state) |
| Both | Accelerated Pathways (e.g., Breakthrough Therapy, PRIME) | For serious conditions with unmet need. Early & intensive agency guidance, rolling review. | Can reduce development time by ~25-30%. |
Vector-Specific Regulatory Considerations & Preclinical Requirements
Different vector classes present unique safety and manufacturing challenges that shape regulatory expectations.
Table 2: Key Preclinical Study Elements by Vector Class
| Vector Class | Genotoxicity & Insertional Mutagenesis | Immunogenicity & Toxicity | Biodistribution & Shedding | Durability of Expression |
|---|---|---|---|---|
| Retroviral (γ-Retrovirus) | Critical. Long-term follow-up (LTFU) ≥15 years. Oncogenicity studies. | Assess immune response to vector & transgene. | Assess risk to gonads; vector integration site analysis. | Long-term (integrating). |
| Lentiviral | Critical. LTFU ≥15 years. Integration site analysis (non-oncogenic models). | Similar to retroviral; strong focus on VSV-G or other envelope immune response. | Broad tissue distribution; must assess gonads and CNS. | Long-term (integrating). |
| Adenoviral (Ad5, HAdV) | Low concern (non-integrating). | Primary concern. Dose-limiting acute inflammatory toxicity, cytokine storm. Pre-existing immunity studies. | High initial liver tropism; shedding studies critical. | Transient (weeks-months). |
| Adeno-Associated Viral (AAV) | Low concern (predominantly episomal). | Critical. Capsid & transgene-directed immune responses. Hepatotoxicity, thrombotic microangiopathy. | Extensive biodistribution to target & non-target tissues (e.g., DRG). Lifelong shedding possible. | Long-term (persistent episomal). |
| Non-Viral (LNPs, Polymers) | Very low concern. | Reactogenicity (infusion reactions), component-specific toxicity (e.g., ionizable lipid). | Rapid clearance; limited persistence. Shedding not applicable. | Transient (days-weeks). |
Detailed Experimental Protocols for Critical Preclinical Assessments
Protocol 3.1: AAV Vector Biodistribution & Shedding Study (QPCR-based)
Objective: Quantify vector genome (VG) persistence in target/non-target tissues and detect shedding in bodily fluids. Materials: AAV vector stock (titer known); Tissues (e.g., liver, heart, CNS, gonads, etc.); Bodily fluids (saliva, semen, urine, feces); DNeasy Blood & Tissue Kit (Qiagen); TaqMan-based qPCR reagents; primers/probe specific to vector sequence (e.g., polyA signal); digital PCR system (optional for absolute quantification). Methodology:
- Dosing: Administer AAV vector to animal model (e.g., mouse, NHP) via relevant route (IV, IM, ICV).
- Sample Collection: At predetermined timepoints (e.g., 1, 4, 12, 26 weeks), collect tissues (~50mg) and excreta.
- DNA Extraction: Homogenize tissues. Extract total genomic DNA. For fluids, concentrate virus if necessary before extraction.
- qPCR Assay: Prepare standard curve using a plasmid containing the target sequence (10^1 to 10^7 copies/µL). Run samples, standards, and negative controls in triplicate.
- Data Analysis: Calculate VG copies per µg of total genomic DNA (tissues) or per mL (fluids). Report mean ± SD. Shedding is considered positive if levels exceed assay cutoff in two consecutive timepoints.
Protocol 3.2: Lentiviral Vector Integration Site Analysis (LAM-PCR)
Objective: Identify genomic locations of vector proviral integration to assess insertional mutagenesis risk. Materials: Genomic DNA from transduced cells; Restriction enzyme (e.g., MseI); T4 DNA Ligase; Linker cassette; Biotinylated primer complementary to vector LTR; Magnetic streptavidin beads; PCR reagents; Next-generation sequencing (NGS) platform. Methodology:
- Digestion & Ligation: Digest 1µg gDNA with MseI. Ligate a double-stranded linker cassette to the restriction ends.
- Nested PCR:
- Primary PCR: Use a linker-specific primer and a biotinylated LTR primer. Amplify integration fragments.
- Capture: Bind biotinylated products to streptavidin beads.
- Secondary PCR: Perform nested PCR on beads using nested primers to increase specificity.
- Sequencing & Analysis: Purify PCR products and subject to NGS. Map sequence reads to the reference genome using bioinformatics tools (e.g., HISAP, VISPA). Analyze for clusters near oncogenes (Common Integration Sites).
Visualization: Regulatory & Development Workflows
Title: Gene Therapy Product Development Pathway
Title: Primary Safety Concerns by Vector Class
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Preclinical Vector Evaluation
| Reagent/Material | Primary Function | Example Vendor/Product |
|---|---|---|
| HEK293T/HEK293 Cells | Standard cell line for production of lentiviral, retroviral, and AAV vectors (provide necessary adenoviral genes). | ATCC (CRL-3216) |
| Adeno-X 293 Cells | Specifically designed for high-titer adenoviral production; expresses higher levels of Ad5 E1 genes. | Takara Bio (632271) |
| pHelper, pAAV, pRC Plasmids | Triple-transfection system for AAV production. pRC provides rep/cap genes. | Addgene (various), Vigene Biosciences |
| Lenti-X Concentrator | Simplifies lentiviral vector concentration from supernatant via precipitation. | Takara Bio (631231) |
| Iodixanol Density Gradient Media | Purification of AAV vectors by ultracentrifugation; achieves high purity and potency. | Cytiva (OptiPrep, 1030061) |
| Anti-AAV Neutralizing Antibody Assay Kit | Measures pre-existing or treatment-induced neutralizing antibodies against specific AAV serotypes. | Progen (AAV Neutralization Assay) |
| Vector Genome Titer Kit (qPCR) | Absolute quantification of vector genomes (vg/mL) for dose standardization. | Applied Biological Materials (G064) |
| ELISpot Kit (IFN-γ, IL-4) | Assess T-cell immunogenicity against capsid/transgene antigens. | Mabtech, BD Biosciences |
| Next-Generation Sequencing Service | For integration site analysis (LAM-PCR), vector integrity, and biodistribution studies. | Eurofins Genomics, Illumina |
| In Vivo Imaging System (IVIS) | Tracks bioluminescent reporter gene expression for preliminary biodistribution & kinetics. | PerkinElmer |
The translational success of viral and non-viral gene delivery vectors is fundamentally constrained by scalable manufacturing. While basic research optimizes transfection efficiency and tropism, the principles of viral (e.g., AAV, Lentivirus) and non-viral (e.g., LNPs, polymers) vectorology must be evaluated through the lens of production logistics. This guide provides a technical framework for assessing scalability, bridging the gap between bench-top discovery and commercial-scale Good Manufacturing Practice (GMP) production.
Core Scalability Challenges for Vector Platforms
Scalability is not linear. Key challenges differ by vector type.
Table 1: Scalability Profile of Major Gene Delivery Vectors
| Vector Type | Primary Production System | Key Scalability Bottleneck | Typical Maximum Titer (Research-Scale) | Critical Quality Attribute (CQA) Most Affected by Scale-Up |
|---|---|---|---|---|
| Adeno-Associated Virus (AAV) | HEK293 Suspension/Adherent; Sf9/Baculovirus | Plasmid Transfection Efficiency; HSV Co-infection Complexity; Purification of Empty/Full Capsids | 1e14 – 1e15 vg/L (HEK293) | Full/Empty Capsid Ratio; Potency (vg/transducing unit) |
| Lentivirus (LV) | HEK293T Adherent; Suspension-Adapted | Transient Transfection Complexity; Vector Stability | 1e7 – 1e8 TU/mL (Concentrated) | Infectivity; Residual Plasmid DNA |
| Lipid Nanoparticles (LNP) | Microfluidic Mixing (T-junction, etc.) | Mixing Efficiency & Consistency; Lipid Stock Stability | Varies by payload (e.g., 1-10 mg/mL mRNA) | Encapsulation Efficiency (%); Particle Size (PDI); pKa |
| Polymeric Vectors (e.g., PEI) | Solvent Evaporation / Nano-precipitation | Batch-to-Batch Polymer Consistency; Removal of Solvent Residues | Varies by polymer | Polyplex Stability; Charge (Zeta Potential) |
Manufacturing Logistics: From Process to Platform
A future-proof strategy requires mapping the entire workflow, identifying points of failure.
Diagram 1: High-Level Gene Therapy Manufacturing Workflow
Upstream Process Intensification
Protocol 3.1.1: Bench-Scale Suspension AAV Production in HEK293 Cells (Seed Train Model)
- Objective: Generate data predictive of bioreactor performance.
- Materials: HEK293SF suspension cells, PEIpro transfection reagent, three-plasmid system (rep/cap, GOI, helper), chemically defined medium, 125mL-1L shake flasks or bench-top bioreactors.
- Method:
- Seed Train Expansion: Thaw cells and expand through successive passages in shake flasks, maintaining viability >95% and cell diameter within +/-2µm of baseline.
- Transfection: At a defined cell density (e.g., 3e6 cells/mL), co-transfect with three plasmids at optimized ratios (e.g., 1:1:1 mass ratio). Use linear PEIpro (1 µg/µL) at a DNA:PEI ratio of 1:3.
- Harvest Timing: Monitor glucose/lactate and cell viability. Harvest cell slurry 48-72 hours post-transfection.
- Data Collection: Record peak cell density, viability at harvest, plasmid consumption (mg/L), and initial titer via ddPCR. Critical Scaling Parameter: The cell-specific plasmid consumption (mg plasmid/10^6 cells) is a key metric for cost forecasting at commercial scale.
Downstream Purification & Analytics
Protocol 3.1.2: Analytical Ultracentrifugation (AUC) for AAV Empty/Full Capsid Quantification
- Objective: Determine the ratio of genome-containing (full) to empty capsids, a critical quality attribute.
- Materials: Purified AAV sample, reference buffer, Beckman Coulter ProteomeLab XL-A/XL-I, two-channel centerpiece, absorbance optics.
- Method:
- Sample Preparation: Dialyze AAV sample into a suitable buffer (e.g., PBS, 200mM NaCl). Load 400 µL into one channel of a centerpiece; load reference buffer into the opposite channel.
- Run Parameters: Set temperature to 20°C. Use a rotor speed of 10,000-12,000 rpm for sedimentation velocity. Collect absorbance data at 260nm (for DNA in full capsids) and 280nm (for total protein).
- Data Analysis: Use SEDFIT software to model continuous c(s) distributions. The 260nm scan reveals the sedimentation coefficient for DNA-containing capsids (~90-110S); the 280nm scan shows the total capsid population. The ratio of areas under the peaks quantifies the empty/full ratio.
Table 2: Key Analytical Methods for Vector Characterization at Scale
| Analytical Method | Measured Attribute | Impact on Scalability Assessment |
|---|---|---|
| ddPCR / qPCR | Vector Genome Titer (vg/mL) | Determines overall yield efficiency of process step. |
| HPLC-SEC / AUC | Aggregation, Empty/Full Ratio | Indicates purification process robustness and final product quality. |
| Dynamic Light Scattering (DLS) | Particle Size & PDI (for LNPs/Polymers) | Monitors formulation consistency across batch sizes. |
| Residual Host Cell DNA/Protein Assays | Product Purity | Validates clearance capability of purification train at high load. |
| NGS | Vector Identity & Integrity | Ensures genetic fidelity is maintained during scale-up. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Scalability Assessment Experiments
| Item | Function in Scalability Context | Example Vendor/Product |
|---|---|---|
| Suspension-Adapted Cell Lines | Enable direct scale-up from shake flasks to bioreactors without adaptation. | Thermo Fisher Gibco Expi293F; Merck HEK293SF |
| GMP-Grade Transfection Reagents | Provide a chemically defined, animal-origin-free path from research to GMP. | Polyplus PEIpro; Mirus Bio TransIT-VirusGEN |
| Microfluidic Mixing Devices | Allow for scalable, reproducible nanoprecipitation of LNPs/nanoparticles. | Precision NanoSystems Ignite; Cytiva ÅkTA microflow system |
| Process Chromatography Resins | Enable capture and polishing step development with scalable bead chemistry. | Cytiva Capto; Tosoh Butyl- and Heparin- Sepharose |
| Single-Use Bioreactors | Mitigate cross-contamination risk and reduce cleaning validation burden. | Sartorius BIOSTAT STR; Thermo Fisher HyPerforma DynaDrive |
| High-Throughput Analytics | Accelerate process optimization with minimal material use (DoE-friendly). | Wyatt Technology DynaPro Plate Reader (DLS); Lunatic (UV-Vis) |
Diagram 2: Decision Logic for Initial Manufacturing Platform Selection
Future-proofing strategy demands that scalability and manufacturing logistics be incorporated as core design principles from the earliest stages of vector research. By employing scalable production models, rigorous analytical comparability studies, and a deep understanding of the cost and quality drivers inherent to each platform, researchers can de-risk the translational pathway and accelerate the delivery of genetic medicines.
Conclusion
The choice between viral and non-viral gene delivery vectors is not a binary one but a strategic decision based on the specific therapeutic goal, target tissue, required expression kinetics, and safety profile. Viral vectors offer high efficiency and persistence, while non-viral vectors provide greater safety, cargo flexibility, and simpler manufacturing. The future lies in hybrid and smart vector systems that combine the advantages of both platforms. As the fields of gene therapy, mRNA vaccines, and gene editing continue to explode, mastering these fundamental principles is paramount for researchers and developers aiming to translate genetic medicines from promising concepts into clinical realities. Continuous innovation in vector engineering is essential to overcome remaining biological barriers and unlock the full potential of genetic medicine.